중심 원리 변환기 III DNA RNA 단백질을 잇는 해석 가능한 AI
CDT‑III는 두 단계의 Virtual Cell Embedder(VCE‑N, VCE‑C) 구조로 DNA‑RNA‑단백질 흐름을 모델링한다. 전사와 번역을 각각 전용 어텐션 모듈로 구현해 CRISPRi 교란에 대한 mRNA와 표면 단백질 변화를 동시에 예측한다. 5개의 보류 유전자에서 RNA 상관계수 r=0.843, 단백질 r=0.969를 달성했으며, 단백질 예측이 RNA 예측을 향상시키고 DNA 수준의 해석 가능성을 30 % 높였다. CD52 …
저자: Nobuyuki Ota
본 논문은 “Central Dogma Transformer III”(CDT‑III)라는 새로운 인공지능 모델을 제안한다. 기존의 생물학 AI 모델은 주로 전사 단계에서 멈추어 RNA 변화를 예측했으며, 단백질 수준의 표현을 다루지 못해 약물 개발 시 부작용 예측에 한계가 있었다. CDT‑III는 이러한 한계를 극복하고자, DNA‑RNA‑단백질 전 과정을 모델링하는 두 단계의 Virtual Cell Embedder(VCE‑N, VCE‑C) 구조를 설계했다.
VCE‑N은 핵 내 전사를 담당한다. Enformer로부터 얻은 DNA 임베딩(≈115 kb)과 세포별 RNA 발현 데이터를 입력으로, DNA 자체 어텐션(2개)과 RNA 자체 어텐션(1개), 그리고 DNA‑to‑RNA 교차 어텐션을 통해 전사 조절을 학습한다. 이 단계는 기존 CDT‑II와 동일한 구조를 유지해 사전 학습된 가중치를 100 % 전이한다.
VCE‑C는 세포질 번역을 담당한다. VCE‑N에서 생성된 RNA 임베딩을 쿼리로, 표면 단백질(ADT) 표현을 키·밸류로 사용해 RNA‑to‑Protein 교차 어텐션을 수행한다. 여기에는 단백질 자체 어텐션(1개)과 번역 조절을 위한 교차 어텐션이 포함된다. 두 단계의 출력은 각각 RNA와 단백질 예측 헤드에 연결된다.
학습은 두 단계로 진행된다. 첫 번째 단계에서는 VCE‑N을 고정하고 VCE‑C만 학습해 단백질 경로가 초기화되도록 한다. 두 번째 단계에서는 전체 파라미터를 미세 조정하며, 손실 함수 L = L_RNA + 0.1·L_Protein을 최소화한다. L_RNA는 2 361개의 유전자에 대한 MSE, L_Protein은 검출된 65개의 ADT에 대한 MSE를 사용한다. λ=0.1은 RNA와 단백질 손실을 균형 있게 반영하도록 실험적으로 선정된 값이다.
데이터는 K562 세포주에서 수행된 STING‑seq v2(CRISPRi, scRNA‑seq, CITE‑seq) 데이터를 사용했다. 총 8 250개의 교란 세포와 2 078개의 비교 대조군이 포함되었으며, 5개의 유전자를 보류 집합으로 설정해 모델의 일반화 능력을 검증했다.
성능 결과는 다음과 같다. RNA 예측에서는 보류 유전자에 대해 평균 Pearson r=0.843을 기록했으며, 이는 RNA‑only 모델인 CDT‑II(0.804)보다 4.9 % 향상된 수치이다. 단백질 예측에서는 65개의 검출 단백질에 대해 평균 r=0.969를 달성했다. 셀 수준에서는 단백질 r=0.28로 측정되었는데, 이는 ADT 측정 노이즈를 반영한다.
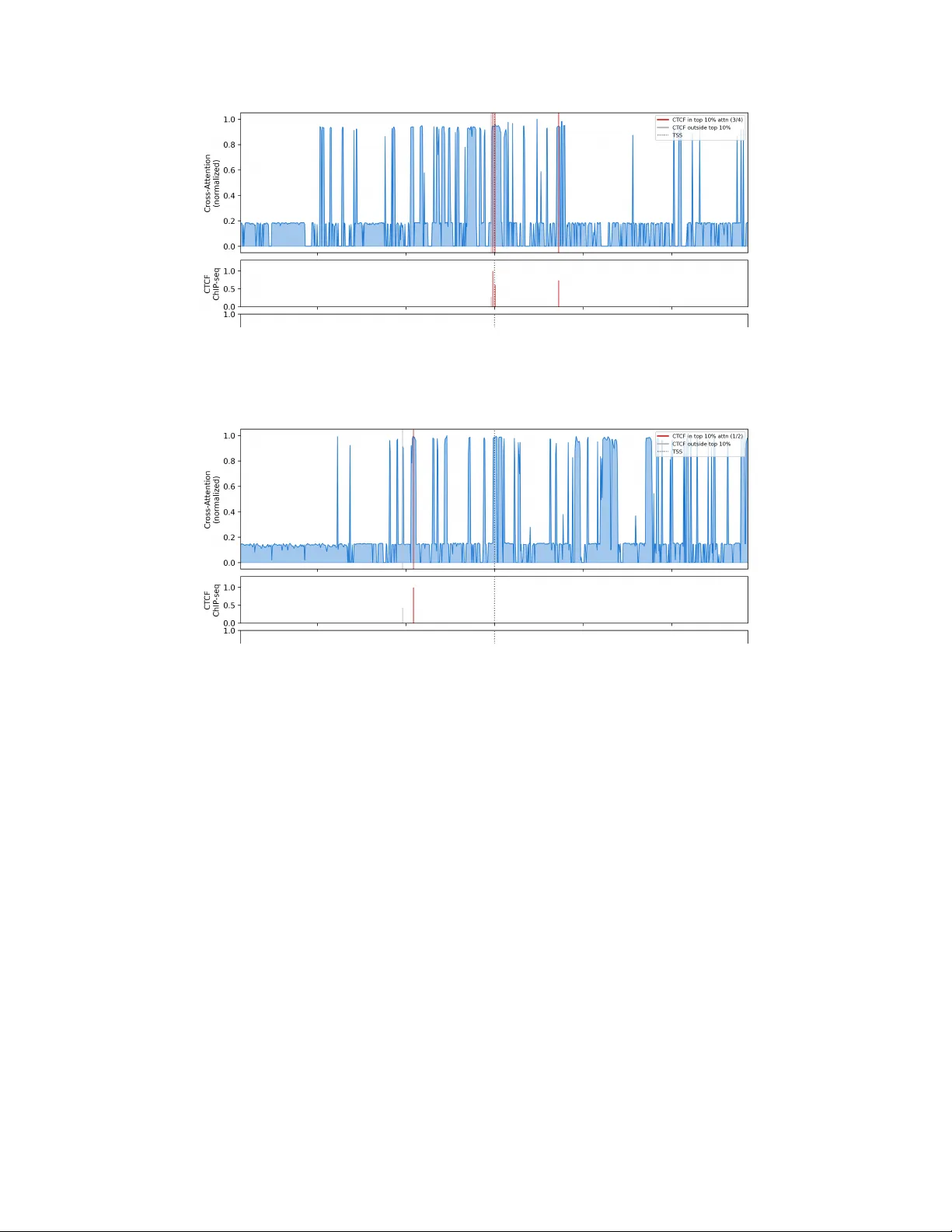
해석 가능성 측면에서는 DNA‑to‑RNA 교차 어텐션이 CTCF 결합 부위에 8.59배(±30 %) 풍부하게 집중되는 것이 확인되었다. 이는 기존 CDT‑II의 6.6배 대비 30 % 향상된 결과이며, 모든 27개의 CTCF 부위가 2배 이상 풍부하게 나타났다(P < 0.001). 또한 Hi‑C 3D 접촉 비율이 평균 1.30배 증가했으며, 이는 모델이 3차원 크로마틴 구조를 암시적으로 학습했음을 의미한다.
특히 단백질 감독이 DNA 수준 해석을 개선한다는 새로운 발견이 있다. 단백질 손실이 VCE‑C를 통해 VCE‑N의 교차 어텐션 가중치에 역전파되면서, 단백질 발현에 결정적인 조절 요소인 CTCF 부위가 더 명확히 강조된다. 이는 다중 과제 학습이 상위 레이어의 표현을 정교화하고, 생물학적 인과 관계를 더 잘 포착하게 만든다.
임상 적용 사례로 CD52 억제(알렘투주맙 모사) 실험을 수행했다. 모델은 29개의 단백질 변화를 모두 올바른 방향으로 예측했으며, 알려진 부작용 7개 중 5개를 재발견했다. 또한 무교란 기반 그래디언트 분석(∂ŷ_Prot,j/∂x_RNA,g)을 통해 r=0.939의 높은 상관을 보이며, 새로운 교란에 대해 실험 없이 부작용 프로파일을 생성할 수 있음을 입증했다. 이를 통해 2 361개의 유전자를 추가 실험 없이 스크리닝할 수 있다.
결론적으로 CDT‑III는 중앙 교리(central dogma)를 구조적으로 모델에 반영함으로써, 예측 정확도와 해석 가능성을 동시에 향상시켰다. 전사·번역 단계별 어텐션을 통해 규제 요소(CTCF, TAD 경계)와 3D 크로마틴 구조를 자동으로 탐지하고, RNA‑only 모델이 놓치는 단백질 수준 정보를 포착한다. 향후 다양한 세포 유형과 풍부한 단백질 데이터셋에 적용한다면, 약물 안전성 평가와 맞춤형 치료 설계에 핵심 도구로 활용될 전망이다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기