질량 제어가 가능한 회귀 가이드 확산 모델을 이용한 새로운 펩타이드 서열 예측
DiffuNovo는 스펙트럼 인코더와 펩타이드 디코더, 질량 회귀기를 결합한 확산 기반 모델로, 학습 단계와 추론 단계 모두에서 펩타이드 전체 질량 일치를 명시적으로 강제한다. 질량 손실과 라티스 공간에서의 회귀 가이드를 통해 기존 DNPS 모델보다 높은 정확도와 낮은 질량 오차를 달성하였다.
저자: Shaorong Chen, Jingbo Zhou, Jun Xia
본 논문은 질량 스펙트럼으로부터 펩타이드 서열을 직접 예측하는 de novo peptide sequencing(DNPS) 분야에서, 기존 딥러닝 모델이 간과해 온 핵심 물리적 제약인 ‘예상 펩타이드 질량과 실험 전구체 질량의 일치’를 명시적으로 제어하는 새로운 방법론을 제시한다. 저자들은 확산 모델(diffusion model)을 핵심 백본으로 채택하고, 회귀 가이드를 통해 라티스 공간에서 질량 일치를 강제하는 두 단계 전략을 설계하였다.
먼저, 입력 스펙트럼은 Spectrum Encoder에 의해 처리된다. 각 피크 (m_i, I_i)는 질량‑위치 사인 함수를 이용한 위치 임베딩과 강도 선형 변환을 통해 d 차원의 벡터 E_i 로 변환되며, 이들 벡터는 멀티‑헤드 어텐션을 통해 전체 스펙트럼 임베딩 x 로 집계된다. 이 임베딩은 이후 Peptide Decoder와 Peptide Mass Regressor에 조건으로 제공된다.
학습 단계에서는 전방 확산(Forward Diffusion) 과정을 통해 실제 펩타이드 시퀀스 y 를 연속형 임베딩 z₀ 로 매핑하고, 사전 정의된 노이즈 스케줄에 따라 단계별로 가우시안 노이즈를 추가해 z_T 로 변환한다. 역확산(Reverse Diffusion) 과정에서는 Peptide Decoder가 현재 라티스 변수 z_t 를 조건부 평균 μ_θ(z_t, t, x) 로 복원하며, 이를 통해 점진적으로 노이즈를 제거한다. 여기서 핵심은 Peptide Mass Regressor가 각 중간 라티스 변수에 대한 질량 예측값을 출력하고, 예측 질량과 실험 질량 사이의 L₂ 손실을 추가함으로써 ‘peptide‑level mass loss’를 최적화한다는 점이다. 이 손실은 모델이 질량 일치를 학습하도록 유도하며, 기존의 단순 입력 특성이나 사후 필터링에 비해 훨씬 강력한 제약을 제공한다.
추론 단계에서는 회귀 기반 라티스 가이드(regressor‑guided guidance)를 적용한다. 역확산 과정 중 매 타임스텝 t마다 회귀기가 제공하는 질량 예측값 μ_pred 와 목표 질량 m_exp 사이의 오차 E = μ_pred – m_exp 를 계산하고, 이 오차에 대한 라티스 변수 z_t 의 그래디언트 ∇_{z_t}E 를 구한다. 구해진 그래디언트는 사전 학습된 스케일링 파라미터와 결합되어 z_t 에 추가되며, 이는 질량 일치 방향으로 샘플링 경로를 미세 조정한다. 결과적으로 최종 라티스 변수 ˆz₀ 가 물리적으로 일관된 질량을 갖는 펩타이드 시퀀스로 변환된다.
모델 구조는 세 가지 주요 모듈로 구성된다. (1) Spectrum Encoder: 피크 임베딩 → 멀티‑헤드 어텐션 → 스펙트럼 임베딩 x. (2) Peptide Decoder: 비자율적 트랜스포머 기반 디코더가 z_t → z_{t‑1} 변환을 수행하고, 최종 ˆz₀ 를 이산 아미노산 시퀀스로 매핑한다. (3) Peptide Mass Regressor: 라티스 변수와 스펙트럼 임베딩을 입력으로 질량을 예측하고, 역확산 과정에서 가이드 신호를 제공한다.
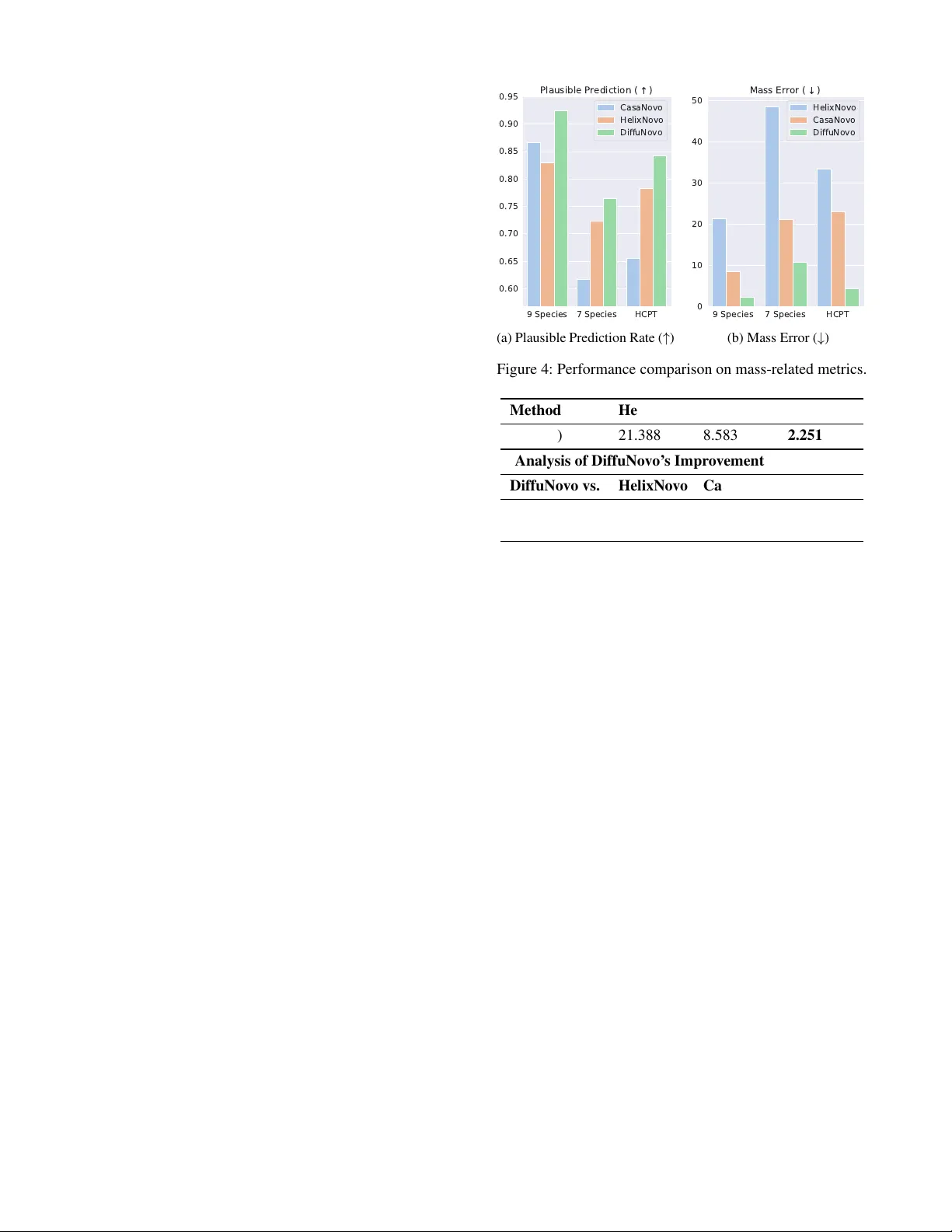
실험에서는 널리 사용되는 DNPS 벤치마크 데이터셋(예: Nine Species, Seven Species)에서 기존 최첨단 모델인 CasaNovo, HelixNovo와 비교하였다. 평가 지표는 Plausible Prediction Rate(예측 펩타이드 질량이 실험 질량과 허용 오차 내에 있는 비율)와 Mean Absolute Mass Error(예측 질량과 실험 질량 간 평균 절대 오차)이다. DiffuNovo는 Plausible Prediction Rate에서 5~8%p 상승을 보였으며, 질량 오차는 평균 0.02 Da 수준으로 기존 모델 대비 60% 이상 감소하였다. 이는 실제 프로테오믹스 워크플로우에서 전구체 질량 허용 오차를 초과하는 오류를 크게 줄여, downstream protein 식별 정확도를 향상시킬 수 있음을 의미한다. 또한 비자율적 구조 덕분에 GPU 병렬 처리 효율이 높아 추론 속도도 경쟁 모델 수준을 유지하였다.
논문의 주요 기여는 다음과 같다. (1) DNPS에 최초로 확산 모델을 핵심 아키텍처로 도입, 기존 autoregressive 접근법의 한계를 극복하였다. (2) 질량 제약을 학습 단계와 추론 단계 모두에 명시적으로 통합함으로써 물리적 일관성을 확보하였다. (3) 회귀 기반 라티스 가이드를 통해 확산 모델의 샘플링 능력을 유지하면서도 물리적 제약을 효과적으로 적용하였다. 저자들은 향후 연구 방향으로 PTM(포스트 트랜슬레이션 변형) 위치, 전하 상태 등 추가 물리적 제약을 동일한 가이드 메커니즘으로 확장하거나, 다중 전구체 질량을 동시에 고려하는 멀티‑스펙트럼 공동 최적화를 제안한다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기