Regressor-guided Diffusion Model for De Novo Peptide Sequencing with Explicit Mass Control
The discovery of novel proteins relies on sensitive protein identification, for which de novo peptide sequencing (DNPS) from mass spectra is a crucial approach. While deep learning has advanced DNPS, existing models inadequately enforce the fundament…
Authors: Shaorong Chen, Jingbo Zhou, Jun Xia
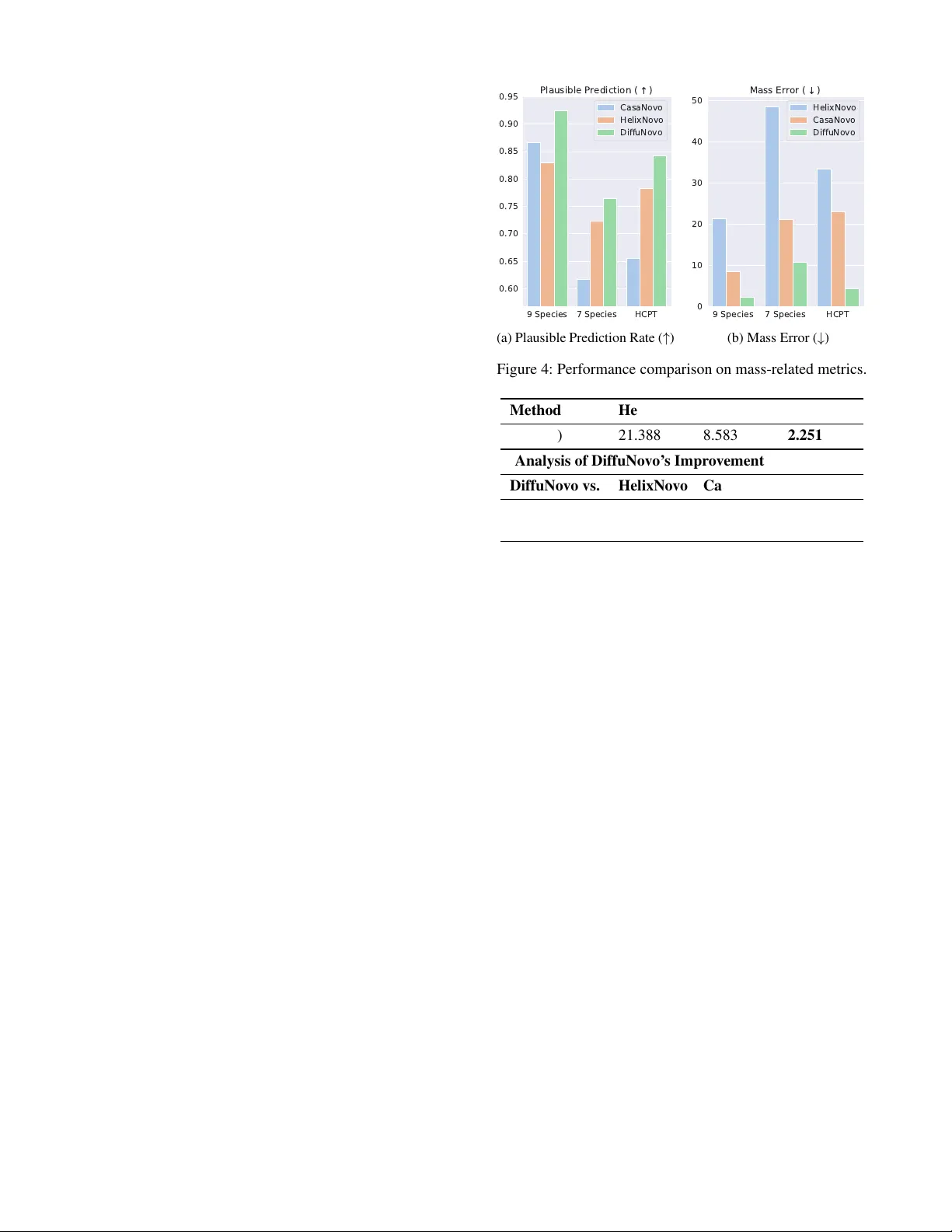
Regr essor -guided Diffusion Model f or De Novo P eptide Sequencing with Explicit Mass Control Shaorong Chen 1,2 , Jingbo Zhou 1,2 , Jun Xia 3,4 † 1 Zhejiang Univ ersity , Hangzhou, China, 310058 2 AI Lab, W estlake Uni versity , Hangzhou, China, 310030 3 AIMS Lab, The Hong K ong University of Science and T echnology (Guangzhou), Guangzhou, China, 511453 4 The Hong K ong University of Science and T echnology , Hong K ong, China, 999077 chenshaorong@westlake.edu.cn, junxia@hkust-gz.edu.cn Abstract The discov ery of nov el proteins relies on sensitiv e protein identification, for which de no vo peptide sequencing (DNPS) from mass spectra is a crucial approach. While deep learn- ing has adv anced DNPS, existing models inadequately en- force the fundamental mass consistency constraint—that a predicted peptide’ s mass must match the experimental mea- sured precursor mass. Pre vious DNPS methods often treat this critical information as a simple input feature or use it in post-processing, leading to numerous implausible predic- tions that do not adhere to this fundamental physical prop- erty . T o address this limitation, we introduce DiffuNov o, a nov el regressor-guided diffusion model for de novo peptide sequencing that provides explicit peptide-level mass control. Our approach integrates the mass constraint at tw o critical stages: during training, a nov el peptide-le vel mass loss guides model optimization, while at inference, re gressor-based guid- ance from gradient-based updates in the latent space steers the generation to compel the predicted peptide adheres to the mass constraint. Comprehensi ve ev aluations on established benchmarks demonstrate that DiffuNo vo surpasses state-of- the-art methods in DNPS accuracy . Additionally , as the first DNPS model to employ a diffusion model as its core back- bone, Dif fuNovo lev erages the powerful controllability of dif- fusion architecture and achiev es a significant reduction in mass error , thereby producing much more physically plau- sible peptides. These innov ations represent a substantial ad- vancement to ward robust and broadly applicable DNPS. The source code is av ailable in the supplementary material. 1 Introduction The identification of the complete set of proteins—the pro- teome—within a biological sample is a fundamental task in biomedicine. A comprehensiv e understanding of this task is critical for elucidating disease mechanisms (Aebersold and Mann 2016), discovering biomarkers (Geyer et al. 2017), and identifying nov el therapeutic targets for drug dev el- opment (Moll and Colombo 2019). The principal high- throughput technology for large-scale protein analysis is tan- dem mass spectrometry (Aebersold and Mann 2003), which is reno wned for its high sensitivity and specificity in charac- terizing complex biological mixtures and has re volutionized the way we study proteins on a large scale. As depicted in Copyright © 2026, Association for the Advancement of Artificial Intelligence (www .aaai.org). All rights reserved. GASMNT ... PEPTED... P E P T E D E P G A S M N T Protein Samples Mixture of Peptides Mass Spectrometer Mass Spectrum Peptide Sequence Protein Sequence Digestion DNPS Assembly Analysis Figure 1: Schematic of a typical bottom-up proteomics workflo w (Zhang et al. 2013). Proteins are digested into pep- tides and analyzed by mass spectrometer to produce mass spectrum. DNPS is the process of inferring a peptide’ s se- quence directly from its spectrum. The resulting peptide se- quences can then be used for protein sequence assembly . Figure 1, the standard workflo w (Zhang et al. 2013) begins with the enzymatic digestion of proteins from a sample into a mixture of smaller , more analytically tractable molecules called peptides. These peptides are then separated and intro- duced into a mass spectrometer for a two-stage analysis. In the first stage, the mass-to-charge ratio of the intact peptide, precursor ion, is measured. In the second stage, this precur- sor ion is isolated and fragmented, and the mass-to-charge ratios of the resulting fragment ions are measured to gen- erate a mass spectrum, which serves as a fingerprint of the original peptide. The central computational challenge in this workflo w is solving an in verse problem: determin- ing the amino acid sequence of a peptide fr om its pre- cursor information and mass spectrum. Successfully de- ciphering this mass spectrum is the crucial step that enables protein identification from the sample. T o this end, de nov o peptide sequencing (DNPS) offers a database-independent paradigm. It directly interprets the mass spectrum to deduce the peptide sequence from first principles, circumventing the need for a existing database. Conceptually , this task is analo- gous to sequence-to-sequence tasks (Sutske ver , V inyals, and Le 2014) in artificial intelligence, such as machine transla- tion. In this analogy , the mass spectrum acts as the input (like a source language), which the model must translate into the target output of a peptide sequence (lik e a target language). 9 Species 7 Species HCPT 0.60 0.65 0.70 0.75 0.80 0.85 0.90 0.95 P l a u s i b l e P r e d i c t i o n ( ) CasaNovo HelixNovo DiffuNovo (a) Plausible Prediction Rate ( ↑ ) 9 Species 7 Species HCPT 0 10 20 30 40 50 M a s s E r r o r ( ) HelixNovo CasaNovo DiffuNovo (b) Mass Error ( ↓ ) Figure 2: Performance comparison of DiffuNo vo with state- of-the-art methods on mass-related metrics. Plausible pre- diction rate is the percentage of predicted peptides whose mass satisfies the e xperimental mass (higher is better). Mass error is the mean absolute error between the predicted pep- tide mass and the experimental mass (lo wer is better). Deep learning has been widely applied in computational biology , and a series of deep learning-based DNPS mod- els (T ran et al. 2017; Qiao et al. 2021; Y ilmaz et al. 2022; Mao et al. 2023; Xia et al. 2024) hav e demonstrated signif- icant progress. Despite these adv ances, existing models fail to suf ficiently le verage a ke y principle: the mass consistency constraint. This constraint dictates that the theoretical mass of a predicted peptide sequence must match the experimen- tally measured precursor mass within a small tolerance. Pre- vious methods typically frame DNPS as a multi-label clas- sification problem, training an autoregressiv e model with an amino-acid-lev el loss function (e.g., cross-entropy). W ithin this framew ork, the precursor mass is often handled sub- optimally: it is either treated as just another numerical in- put feature or used merely as a post-processing filter to dis- card inv alid candidates. This inadequate enforcement of the mass constraint leads to numerous implausible predictions. Consequently , existing DNPS models often fall short in pre- dicting peptides that are plausible with respect to their ex- perimental measured mass. Therefore, de veloping a method that lev erages powerful generati ve models like diffusion to enforce this mass constraint is crucial for advancing DNPS. T o address this limitation, we introduce Dif fuNovo, a nov el regressor-guided diffusion model for de nov o peptide sequencing that provides explicit, peptide-lev el mass con- trol. Dif fuNov o is built on a non-autoregressi ve T ransformer architecture and comprises three main modules: a Spectrum Encoder , a Peptide Decoder , and a Peptide Mass Regressor . The Spectrum Encoder encodes the input mass spectrum into embeddings. Conditioned on this spectral embedding, the Peptide Decoder operates during the rev erse diffusion process to iterati vely denoise a Gaussian noise vector , pass- ing through a series of intermediate latent variables to ulti- mately produce a clean latent representation of the predicted peptide sequence. The core innovation lies in the Peptide Mass Regressor , which guides the intermediate latent v ari- ables throughout this reverse process to enforce mass consis- tency . Specifically , we inte grate the mass constraint at two critical stages: during training, a nov el peptide-le vel mass objectiv e is introduced to train the Regressor to predict the mass corresponding to intermediate latent variables; during inference, the pre-trained Regressor provides guidance to the Peptide Decoder by steering the generation process with gradient-based updates applied to the intermediate latent space. This compels that the final predicted peptide’ s mass adheres to the mass constraint. Comprehensi ve ev aluations on established benchmarks demonstrate that Dif fuNovo sur- passes state-of-the-art methods in DNPS accurac y . More im- portantly , as shown in Figure 2, DiffuNo vo achiev es a signif- icant reduction in mass error compared to baseline models and thereby producing more physically plausible peptides. These innov ations represent a substantial adv ancement to- ward reliable and broadly applicable DNPS. In summary , our core contributions are as follo ws: • As the first DNPS model to feature explicit mass control, DiffuNo vo effecti vely imposes this critical mass con- straint throughout both the training and inference stages. • W e propose DiffuNo vo, a no vel re gressor-guided diffu- sion model for de novo peptide sequencing that provides explicit mass control. T o our knowledge, it is the first DNPS model to utilize diffusion as its core architecture. • Comprehensive ev aluations demonstrate that Dif fuNovo achiev es state-of-the-art DNPS accuracy and, through Regressor guidance, significantly reduces mass error to predict more physically plausible peptides (Figure 2). 2 Related W orks De Nov o Peptide Sequencing (DNPS) With the advent and prosperity of deep learning, a new wa ve of DNPS meth- ods has emerged, achie ving significant performance gains. DeepNov o (T ran et al. 2017) was a pioneering work that first applied deep neural netw orks to DNPS. Subsequent research has le veraged a variety of advanced architectures, includ- ing Geometric Deep Learning (Qiao et al. 2021; Mao et al. 2023), and Transformer architecture (Y ilmaz et al. 2022; Xia et al. 2024). Notably , while InstaNovo+ (Eloff et al. 2023) utilized a dif fusion model for the refinement of predicted peptides, it was employed only as a post-processing step in- stead of the core backbone. T o our kno wledge, Dif fuNov o is the first DNPS model to use diffusion as its core architecture. Diffusion Models for Controllable Generation Diffusion models hav e emerged as a prominent class of generativ e models, renowned for their ability to synthesize high-fidelity and fine-grained controllable samples. Recent studies hav e demonstrated their remarkable performance, not only in continuous domains (Rombach et al. 2022; Ho et al. 2022; Liu et al. 2023), b ut also in discrete domains (Li et al. 2022; Xu et al. 2022; W atson et al. 2023). Several distinct strate- gies have been established to implement controllability , in- cluding: incorporating feedback from an external function (Dhariwal and Nichol 2021), training the dif fusion model to accept conditioning prompt (Rombach et al. 2022), and di- rectly modifying the denoising predictions (Zhang, Rao, and Agrawala 2023). In contrast to the aforementioned works, our research pioneers the application to the DNPS. 3 Preliminary This paper addresses the problem of de novo peptide se- quencing (DNPS), which aims to determine the amino acid sequence of a peptide given its experimental mass spec- trum and precursor information. The mass spectrum is a set of peaks s = { s i } M i =1 = { ( m i , I i ) } M i =1 , where each peak s i = ( m i , I i ) consists of a mass-to-charge ratio m i ∈ R and its corresponding intensity I i ∈ R . The pr ecursor in- formation is a tuple p = ( m prec , c prec ) , where m prec ∈ R is the mass-to-charge of the precursor and c prec ∈ Z + is its charge. The target output is a peptide y , which is a sequence of amino acids y = ( y 1 , y 2 , . . . , y N ) . Each amino acid y i belongs to a pre-defined vocab ulary of amino acids, AA . The number of peaks M and the peptide length N is variable. The experimentally measur ed mass of the peptide m exp is calculated from the precursor p follo wing (Aebersold and Mann 2003). The theor etical mass m pred of predicted pep- tide is the sum of the masses of its constituent amino acids. Formally , the goal of deep learning-based DNPS is learn- ing model with parameters θ that estimates p ( y | s , m exp ; θ ) . 4 Method W e consider the setting of de novo peptide sequencing (DNPS) with explicit mass control. T o render this complex problem more tractable without loss of generality , we de- compose it into two simpler sub-problems: 1) we train a base DNPS model p ( y | s ; θ 1 ) on labeled dataset. 2) for explicit mass control, we train a Regressor , p ( m exp | y ; θ 2 ) , on the same dataset to predict the mass m exp of a peptide sequence y given its high-dimensional latent variable. The goal of Dif- fuNov o is to utilize these two blocks to approximately sam- ple from p ( y | s , m exp , θ ) via Bayes rule: p ( y | s , m exp , θ ) ∝ p ( y | s ; θ 1 ) · p ( m exp | y ; θ 2 ) . (1) Intuitiv ely , the first term p ( y | s ; θ 1 ) encourages the pre- dicted peptides y to be consistent with the mass spectrum s . The second term p ( m exp | y ; θ 2 ) acts as a guidance to compel the predicted sequence y fulfills the mass m exp . The framew ork of our proposed model, DiffuNo vo, is il- lustrated in Figure 3. It comprises three core components based on the T ransformer architecture: a Spectrum En- coder , a Peptide Decoder , and a Peptide Mass Regr essor . First, the Spectrum Encoder generates an embedding of the input mass spectrum s . Conditioned on this embedding, the Peptide Decoder models p ( y | s ; θ 1 ) by progressively de- noising a random Gaussian noise into a latent variable of the peptide sequence during the reverse dif fusion process. W orking in tandem with the Peptide Decoder , the Peptide Mass Re gressor models p ( m exp | y ; θ 2 ) . Re gressor assesses the mass consistency of intermediate latent variables and provides guidance to steer the generation, compelling the fi- nal peptide adheres to the experimental measured mass. 4.1 Encoding of Input by Spectrum Encoder Initially , Dif fuNovo transforms mass spectrum s into a se- quence of high-dimensional vectors { E i } M i =1 , suitable for processing by Transformer -based Spectrum Encoder . W e follow widely used methods (Y ilmaz et al. 2022), inv olves independent vectorization of each peak in mass spectrum. Each peak s i = ( m i , I i ) in the mass spectrum s = { s i } M i =1 is indi vidually mapped to a d -dimensional embed- ding, E i . This transformation is achieved through two paral- lel pathw ays that separately encode the mass-to-charge ratio m i and the intensity I i . The m i is encoded using a sinusoidal positional function, analogous to its use in natural language processing (V aswani et al. 2017), to capture the precise loca- tion of peaks within the mass domain. The intensity is pro- jected into the embedding space via a trainable linear layer W . The final peak embedding E i is the element-wise sum of these two vectors. The formal definitions are as follo ws: E mz i = " sin m i N 1 N 2 d 2 , sin m i N 1 N 4 d 2 , . . . , sin m i N 1 N d d 2 , cos m i N 1 N d +2 d 2 , cos m i N 1 N d +4 d 2 , . . . , cos m i N 1 N 2 2 # (2) E I i = W I i (3) E i = E I i + E mz i (4) where d is the embedding dimension, and W ∈ R d × 1 is a trainable linear layer , N 1 and N 2 are pre-defined scalars. The resulting sequence of peak embeddings, { E i } M i =1 , serves as the input to the Spectrum Encoder . This module employs a multi-head attention mechanism (V asw ani et al. 2017) to compute mass spectrum embedding x , which en- capsulates a holistic representation of the mass spectrum s . Embedding x is then input to the Peptide Decoder . 4.2 T raining Stage of DiffuNovo The training process of our proposed DiffuNo vo operates in two process. The Forward Diffusion Pr ocess is a fixed pro- cedure used during training where, at a giv en timestep, noise is added to the ground-truth peptide embeddings to yield a noisy intermediate latent variable. The Reverse Diffusion Process predicts peptide sequences from this noisy latent variable, conditioned on a mass spectrum embedding. Forward Diffusion Process For the training of Dif- fuNov o, we define a specific forw ard process that constructs a trajectory of latent v ariables, { z t } T t =0 , where T is maxi- mum diffusion timestep. The initial transition from the dis- crete amino acid tokens of y to a continuous latent v ariable z 0 is defined by a conditional distribution: q ϕ ( z 0 | y ) = N ( z 0 ; Emb ϕ ( y ) , (1 − α 0 ) I ) (5) Here, Em b ϕ ( y ) is a learnable function with parameter ϕ that maps the discrete peptide sequence y into a continuous vector , and α 0 is a predefined variance schedule parameter . Subsequently , the v ariable z 0 is gradually perturbed until it conv erges to a standard Gaussian noise. The process at each intermediate timestep t ∈ [1 , T ] can be formalized as: q ( z t | z 0 ) = N ( z t ; √ ¯ α t z 0 , (1 − ¯ α t ) I ) , (6) where, ¯ α t = Q t i =1 α i , and α i is a noise coefficient that de- creases with timestep t , z t is intermediate latent variable. Mass Spectrum Spectrum Embedding Spectrum Encoder (M Layers) Ground T ruth Peptides Peptide Embedding Add Noise P E P T I D E P E P T I D E P E P T I D Forward Diffusion Process Reverse Diffusion Process P E P T I D E P E P T I D E P E P T I D E Predicted Peptides Prediction Head Peptide Decoder (N Layers) Peptide Mass Regressor (L Layers) Figure 3: The architecture of DiffuNo vo. In the Forward Diffusion Process, a ground-truth peptide sequence is con verted into an embedding z 0 and then corrupted by adding noise ov er T timesteps to produce a noisy latent variable z T . DiffuNov o is trained to re verse this process. The Spectrum Encoder transforms the mass spectrum into embedding x . The core of Dif fuNovo is Re verse Dif fusion Process where the Peptide Decoder denoise the noisy latent z t into a cleaner latent z t − 1 conditioned on x . Critically , the Peptide Mass Regressor provides guidance to the Peptide Decoder at each timestep t , compelling the prediction adheres to the mass constraint. Finally , the clean latent ˆ z 0 is passed to a prediction head to output the final predicted peptide. Reverse Diffusion Pr ocess The re verse denoising process is designed to inv ert the forward dif fusion by learning a pa- rameterized transition, p θ ( z t − 1 | z t , t, x ) , that progressively remov es noise conditioned on the mass spectrum embed- ding x . This process starts with a sample z T from a standard Gaussian distribution, z T ∼ N ( 0 , I ) , and iteratively applies the denoising step until t = 0 . Each transition in this re verse Markov chain is modeled as a Gaussian distrib ution: p θ ( z t − 1 | z t , t, x ) = N z t − 1 ; µ θ ( z t , t, x ) , Σ θ ( z t , t, x ) , (7) The v ariance Σ θ is kept fixed to a predefined schedule following (Li et al. 2022). T o parameterizing the mean µ θ , we train the Peptide Decoder denoted as ˆ z 0 = g θ ( z t , t, x ) , to predict the clean embedding ˆ z 0 from the noisy latent v ariable z t . This non-autoregressiv e prediction of the final state is a common and effecti ve strategy in dif fusion models (Lin et al. 2023). The required mean µ θ for Equation 7 can then be calculated in a closed form based on the predicted ˆ z 0 and the current state z t following (Ho, Jain, and Abbeel 2020). The final step of the re verse dif fusion process maps the fully denoised latent variable ˆ z 0 (continuous embedding) to the target peptide sequence y (discrete sequence). This is achiev ed through a prediction head that models the proba- bility of each amino acid in each position i independently: p θ ( y | ˆ z 0 ) = N Y i =1 p θ ( y i | ˆ z i 0 ) (8) The DiffuNov o’ s parameters are optimized by maximizing the evidence lower bound (ELBO) of the log-likelihood (Ho, Jain, and Abbeel 2020). This yields a simplified and ef fec- tiv e training objective, which is a combination of a recon- struction term and a denoising-matching term: L 1 = L Peptide Decoder = E q ϕ ( z 0: T | y ) T X t =1 ∥ z 0 − g θ ( z t , t, x ) ∥ 2 − log p θ ( y | z 0 ) (9) In parallel with the denoising task, we introduce and co- train the Peptide Mass Re gressor . The function of this Re- gressor is to predict the peptide mass m pred directly from the noisy intermediate latent variable z t at any timestep t : m pred ( z t , θ ) = N X i =1 X y j ∈ AA p θ ( y j | z i t , θ ) · m ( y j ) (10) where AA is the set of amino acids and m ( y i ) is the mass of amino acid y i . Regressor is trained on the same dataset as the Peptide Decoder , and its parameters are optimized using mean squared error (MSE) loss between the predicted mass m pred and the experimental measured mass m exp : L 2 = L Regressor = E q ϕ ( z 0: T | y ) ∥ m pred ( z t , θ ) − m exp ) ∥ 2 (11) The whole process of the DiffuNov o model training stage can be summarized by the pseudocode in Algorithm 1. 4.3 Inference Stage of DiffuNo vo Algorithm 1: T raining Stage of DiffuNov o. Input : Labeled Dataset D = { ( s , m exp ) , y } , maximum diffusion timestep T and maximum peptide length N . Output : Optimized model parameters θ . 1: repeat 2: Sample a data instance ( s , m exp , y ) ∼ D . 3: Encode input s = { s i } M i =1 into continuous represen- tations { E i } M i =1 and compute mass spectrum embed- ding: x ← Spectrum Encoder ( { E i } M i =1 ) . 4: Maps the discrete peptide y into embedding vector: z 0 ∼ q ϕ ( z 0 | y ) = N ( z 0 ; Emb ϕ ( y ) , (1 − α 0 ) I ) (12) 5: Sample a timestep t ∼ [1 , T ] and construct the noisy latent variable z t with Gaussian reparameterization: z t ∼ q ( z t | z 0 ) = N ( z t ; √ ¯ α t z 0 , (1 − ¯ α t ) I ) . (13) 6: According to Equation 9 and Equation 11, employ gradient descent to optimize the objectiv e: min θ ( h z 0 − g θ ( z t , t ; x ) 2 − log p θ ( y | z 0 ) i + N X i =1 X y j ∈ AA p θ ( y j | z i t , θ ) · m ( y j ) − m exp 2 ) (14) 7: until conv erged Reverse Pr ocess The inference process of Dif fuNovo is summarized in Algorithm 2. During inference, Dif fuNovo ex ecutes the rev erse diffusion process to predict peptide se- quence y conditioned on input mass spectrum x . This pro- cess begins with an initial Gaussian noise vector z T , which is iterati vely refined through T timesteps. At each step t , the Peptide Decoder tak es the noisy latent variable z t and the spectrum embedding x from the Spectrum Encoder as input to predict a cleaner latent variable z t − 1 : p θ z t − 1 | z t , t, x , m exp ∼ N z t − 1 ; µ θ ( z t , x , t ) + s ∆ z t i , σ I (18) Crucially , the inference process is dif ferent from the re- verse diffusion mentioned in Section 4.2 by: 1) the denoising process begins not by adding noise to a ground-truth peptide y , but by sampling an initial latent variable z T from a stan- dard Gaussian distrib ution. 2) incorporating an explicit mass control by intermediate latent v ariable by the term s ∆ z t i ( s is a scalar) using the trained Peptide Mass Regressor . Our approach to e xplicit mass control is inspired by Equa- tion 1, but instead of directly controlling the discrete peptide sequence, we control the sequence of continuous intermedi- ate latent variables z 0: T during re verse diffusion process. As a further refinement of the simplified formulation in Equa- tion 1, controlling z 0: T is equi valent to decoding from the posterior p ( z 0: T | x , m exp ) = Q T t =1 p ( z t − 1 | z t , x , m exp ) , and we decompose this complex inference problem by: p ( z t − 1 | z t , x , m exp ) ∝ p ( z t − 1 | z t , x ) · p ( m exp | z t − 1 , z t ) (19) Algorithm 2: Inference Process of DiffuNo vo. Input : Inference instance ( s , m exp ) from test dataset D = { ( s , m exp ) } , maximum diffusion decoding timestep T , trained model parameters θ and gradient-based guidance step s , scalar co- efficient λ 1 and λ 2 . Output : Predicted peptide sequence ˆ y . 1: Encode inputs s = { s i } M i =1 into continuous representa- tions { E i } M i =1 and compute mass spectrum embedding: x ← Spectrum Encoder ( { E i } M i =1 ) . 2: Uniformly select a decreasing subsequence of timesteps t M :0 ranging from T to 0 . 3: Sample z t M ∼ N ( 0 , I ) . 4: for i = M to 1 do 5: Get the current timesteps t i and the subsequent timestep t i − 1 from the pre-defined timesteps t M :0 6: Compute denoising mean through Peptide Decoder: µ θ ( z t i , t i , x ) ← λ 1 z t i + λ 2 g θ ( z t i , t i , x ) (15) 7: Compute gradient-based update for latent variables z t i through Peptide Mass Regressor: ∆ z t i = ∇ z t i m pred ( z t i , θ ) − m exp 2 (16) 8: The subsequent latent variables z t i − 1 is then sampled from Gaussian distribution: p θ z t i − 1 | z t i , t i , x , m exp ∼ N z t i − 1 ; µ θ ( z t i , t i , x ) + s ∆ z t i , σ I (17) 9: end for 10: Map z 0 to the peptide sequence ˆ y through prediction head. W e further simplify p ( m exp | z t − 1 , z t ) = p ( m exp | z t − 1 ) via conditional independence assumptions from prior work on controlling dif fusions (Song et al. 2020). Consequently , for the t -th timestep, we run gradient update on z t − 1 : ∇ z t − 1 p ( z t − 1 | z t , x , m exp , θ ) = ∇ z t − 1 p ( z t − 1 | z t , x , θ 1 ) + ∇ z t − 1 p ( m exp | z t − 1 , θ 2 ) where both p ( z t − 1 | z t , x , θ 1 ) and p ( m exp | z t − 1 , θ 2 ) are differentiable: the first term is parametrized by Peptide Decoder , and the second term is parametrized by a neural network-based Peptide Mass Regressor . W e run gradient up- dates ∇ z t − 1 p ( m exp | z t − 1 , θ 2 ) on the latent space to steer it tow ards fulfilling the mass consistency constrain. Final Pr ediction Finally , prediction head maps the fully denoised ˆ z 0 to the predicted peptide sequence y . Similarly to beam search (Freitag and Al-Onaizan 2017), Dif fuNov o predicts a set of candidates and select the final prediction by: • DiffuNovo (Logits): This variant selects the candidate with the highest peptide-level log-probability , as deter- mined by the logits from the final projection head. • DiffuNovo (MBR): This v ariant employs Minimum Bayes Risk (MBR) decoding (Kumar and Byrne 2004) to select the optimal candidates from the predicted set. 5 Experiments 5.1 Experimental Settings All experimental settings in this paper adhere to the Nov oBench benchmark (Zhou et al. 2024). Our ev aluation lev erages three representative datasets, selected for their di- verse sizes, resolutions, and biological origins: the Nine- species Dataset (T ran et al. 2017), the HC-PT Dataset (Elof f et al. 2023), and the Se ven-species Dataset (T ran et al. 2017). T o comprehensi vely e valuate the performance of Dif- fuNov o, we compare it against a suite of advanced baselines, including DeepNov o (Tran et al. 2017), PointNov o (Qiao et al. 2021), CasaNovo (Y ilmaz et al. 2022), AdaNovo (Xia et al. 2024), and HelixNovo (Y ang et al. 2024). The per- formance of all models was assessed using standard met- rics: (1) Peptide-level Precision serves as the primary indi- cator of model performance; (2) Peptide-lev el Area Under the Curve (A UC) assesses performance across different con- fidence thresholds; and (3) Amino Acid-level Precision and Recall ev aluate performance at a finer granularity . 5.2 Experimental Results DiffuNovo Achieves State-of-the-art Performance on Most Benchmark Metrics The empirical results, sum- marized in T able 2 and 3, demonstrate that DiffuNo vo achiev es state-of-the-art performance, consistently outper- forming leading baselines across most datasets and metrics. The performance on standard DNPS benchmarks is de- tailed in T able 2. Our model demonstrates exceptional ca- pabilities, particularly in peptide-level precision. The Dif- fuNov o(Logits) variant achiev es the best peptide precision across the major datasets, with a precision of 0.572 on the 9- species dataset and 0.485 on the HC-PT dataset, decisiv ely outperforming all baseline models. Furthermore, the Dif- fuNov o(MBR) v ariant showcases the model’ s comprehen- siv e po wer by securing top performance across other met- rics, including the highest peptide-lev el A UC and amino acid-lev el precision and recall. These results highlights the DiffuNo vo’ s core strength in accurately predicting the cor- rect peptide sequence for de nov o peptide sequencing. The identification of amino acids with post-translational modifications (PTMs) holds important biological signifi- cance because it plays a pi votal role (Deribe, Pa wson, and Dikic 2010). As detailed in T able 3, DiffuNo vo achie ves the highest PTM precision, outperforming other methods by a substantial margin. By enforcing a strict adherence to the e x- perimental mass through Regressor , DiffuNo vo ef fectiv ely eliminating candidate peptides that are physically implausi- ble. This constraint is crucial for PTMs, where subtle mass shifts differentiate PTM from canonical amino acid. The Peptides Predicted by DiffuNovo Exhibit A Highly Significant Enhancement in Mass Consistency T o quan- titativ ely ev aluate the ef fectiveness of our proposed explicit mass control, we compared the theoretical mass of the pep- tides predicted by DiffuNov o with their experimentally de- termined precursor mass. T able 1 presents the Mean Abso- lute Error of this mass discrepancy . Figure 4 illustrates the plausible prediction rate, defined as the proportion of pre- dictions where the mass error is less than 1e-3 Da. 9 Species 7 Species HCPT 0.60 0.65 0.70 0.75 0.80 0.85 0.90 0.95 P l a u s i b l e P r e d i c t i o n ( ) CasaNovo HelixNovo DiffuNovo (a) Plausible Prediction Rate ( ↑ ) 9 Species 7 Species HCPT 0 10 20 30 40 50 M a s s E r r o r ( ) HelixNovo CasaNovo DiffuNovo (b) Mass Error ( ↓ ) Figure 4: Performance comparison on mass-related metrics. Method HelixNovo CasaNovo DiffuNovo MAE( ↓ ) 21.388 8.583 2.251 Analysis of DiffuNovo’ s Improvement DiffuNovo vs. HelixNo vo CasaNov o DiffuNovo X% Decrease 89.5% 73.8% 0% Fold Decrease 9.50 × 3.81 × 1.00 × T able 1: Evaluation of mass error . W e compares DiffuNo vo with two baselines and reports the Mean Absolute Error (MAE), where lower is better . The upper section shows the mass errors. The lower section quantifies this impro vement. As T able 1, DiffuNo vo achie ves a MAE of 2.251, rep- resenting a remarkable 89.5% (a 9.50-fold decrease) and 73.8% (a 3.81-fold decrease) reduction compared to He- lixNov o and CasaNovo. This empirical evidence strongly validates that the e xplicit mass control is highly effecti ve. As illustrated in Figure 4, DiffuNo vo consistently out- performs state-of-the-art baselines on ke y mass-related met- rics. Figure 4a shows that our model achieves a substantially higher plausible prediction rate, indicating that a greater pro- portion of prediction are physically plausible as their the- oretical mass aligns with the experimental mass. Concur- rently , Figure 4b reveals a dramatic reduction in mass error . This enhanced performance is not coincidental but is in- trinsically linked to the core design of DiffuNo vo: by inte- grating a Regressor to guide in the latent space, our model activ ely steers the dif fusion process towards peptides whose theoretical mass aligns with the experimental mass. The Significant Reduction in Mass Error is Directly At- tributable to the Regressor -based Guidance T o validate the ef fectiveness of Regressor for reduction in mass error , we analyzed the mass error trajectory throughout the re- verse dif fusion process. Figure 5 illustrates three variants of Dif fuNovo: a baseline version without Regressor (Base), and two guided versions with different guidance steps (Dif- fuNov o 1 with step=5e-3 and DiffuNov o 2 with step=1e-2). Peptide-lev el Performance Amino Acid-lev el Performance Models 9-species HC-PT 7-species 9-species HC-PT 7 species Prec. A UC Prec. A UC Prec. A UC Prec. Recall Prec. Recall Prec. Recall DeepNov o 0.428 0.376 0.313 0.255 0.204 0.136 0.696 0.638 0.531 0.534 0.492 0.433 PointNov o 0.480 0.436 0.419 0.373 0.022 0.007 0.740 0.671 0.623 0.622 0.196 0.169 CasaNov o 0.481 0.439 0.211 0.177 0.119 0.084 0.697 0.696 0.442 0.453 0.322 0.327 AdaNov o 0.505 0.469 0.212 0.178 0.174 0.135 0.698 0.709 0.442 0.451 0.379 0.385 HelixNov o 0.517 0.453 0.356 0.318 0.234 0.173 0.765 0.758 0.588 0.582 0.481 0.472 DiffuNo vo(Logits) 0.572 0.413 0.485 0.324 0.233 0.104 0.785 0.783 0.648 0.648 0.430 0.428 DiffuNo vo(MBR) 0.565 0.536 0.458 0.434 0.193 0.162 0.791 0.789 0.654 0.654 0.437 0.435 T able 2: The comparison of de novo peptide sequencing performance between our proposed model, Dif fuNovo, and other state- of-the-art methods on the three benchmark datasets. W e report precision and A UC at the peptide le vel, and precision and recall at the amino acid lev el. The best and the second best are highlighted with bold and underline, respectiv ely . PTM Precision Models Nine-Species HC-PT Sev en-Species DeepNov o 0.576 0.626 0.391 PointNov o 0.629 0.676 0.117 AdaNov o 0.652 0.552 0.448 CasaNov o 0.706 0.501 0.360 HelixNov o 0.680 0.568 0.473 DiffuNovo 0.822 0.705 0.515 T able 3. Empirical comparison of PTM identification. W e ev aluate the ability of DiffuNo vo and other models to iden- tify Post-Translational Modifications (PTMs) on the bench- mark datasets. The best results and the second best are high- lighted with bold and underline, respecti vely . Nor malized T imestep 0 2000 4000 6000 8000 10000 12000 Mass L oss Base DiffuNovo 1 DiffuNovo 2 Figure 5: Mass error loss curv es. The figure illustrates the mass error loss (MSE) as a function of the normalized dif- fusion timestep. W e conducted experiment on 5 random batches (2560 samples), calculating and plotting the loss for each batch (dashed line) and the av erage loss (solid line). The results provide compelling evidence that the regressor -based guidance is a critical component for achie v- ing reduction in mass error . As depicted by the blue curve, the original unguided model reduces the mass error gradu- ally , but its final error remains substantial. In stark contrast, the regressor-based v ariants leads to a markedly low error . Both DiffuNov o 1 (orange curve) and Dif fuNovo 2 (green curve) demonstrate a significantly faster and deeper reduc- tion in mass error from the very early stages of the rev erse diffusion process. This visually confirms that the Regressor effecti vely steers the diffusion tow ards states that are consis- tent with the target e xperimental measured mass. Ablation Study In the ablation study , we inv estigated the contribution of each key component within DiffuNo vo. W e remov ed the Spectrum Encoder , the Peptide Decoder , and the Peptide Mass Re gressor, then trained and e valuated these ablated models on the HC-PT dataset. All other settings were k ept identical. The results is summarized in T able 4, lead to the conclusion that each module is crucial. Model Perf ormance Mass Constraint Peptide AA Plausible MAE Prec.( ↑ ) Prec.( ↑ ) Rate( ↑ ) V alue( ↓ ) Full Model 0.485 0.648 0.843 4.350 - w/o Encoder 0.104 0.313 0.686 16.509 - w/o Decoder 0.066 0.267 0.652 35.260 - w/o Regressor 0.437 0.641 0.703 14.555 T able 4: Ablation study of the model components. 6 Conclusion In this paper , we identify that previous DNPS methods often handle mass information in trivial manner , leading to im- plausible predictions that are inconsistent with the experi- mental mass. T o address this limitation, we introduce Dif- fuNov o, a novel regressor -guided dif fusion model that pro- vides explicit mass control. Guidance from Re gressor via gradient-based updates in the latent space compels the pre- dicted peptides adhere to the mass constraint. Comprehen- siv e ev aluations demonstrate that Dif fuNovo surpasses state- of-the-art methods in DNPS accurac y . W e sho w that the Re- gressor’ s guidance significantly reduces the mass error and increases the rate of plausible predictions. These innovations demonstrate that Dif fuNovo model has achie ved a substan- tial advancement to ward more rob ust and reliable DNPS. 7 Acknowledgments W e sincerely thank the anonymous revie wers for their in- sightful comments and constructiv e suggestions. This re- search is supported by National Natural Science F oundation of China Project (No. 623B2086), T eleAI of China T elecom, T encent and Ant Group References Aebersold, R.; and Mann, M. 2003. Mass spectrometry- based proteomics. Natur e , 422(6928): 198–207. Aebersold, R.; and Mann, M. 2016. Mass-spectrometric exploration of proteome structure and function. Natur e , 537(7620): 347–355. Deribe, Y . L.; Pawson, T .; and Dikic, I. 2010. Post- translational modifications in signal inte gration. Natur e structural & molecular biology , 17(6): 666–672. Dhariwal, P .; and Nichol, A. 2021. Dif fusion models beat gans on image synthesis. Advances in neur al information pr ocessing systems , 34: 8780–8794. Eloff, K.; Kalogeropoulos, K.; Morell, O.; Mabona, A.; Jes- persen, J. B.; W illiams, W .; van Beljouw , S. P .; Skwark, M.; Laustsen, A. H.; Brouns, S. J.; et al. 2023. De nov o peptide sequencing with InstaNov o: Accurate, database-free pep- tide identification for large scale proteomics experiments. bioRxiv , 2023–08. Freitag, M.; and Al-Onaizan, Y . 2017. Beam search strategies for neural machine translation. arXiv pr eprint arXiv:1702.01806 . Geyer , P . E.; Holdt, L. M.; T eupser , D.; and Mann, M. 2017. Revisiting biomarker discovery by plasma pro- teomics. Molecular systems biology , 13(9): 942. Ho, J.; Chan, W .; Saharia, C.; Whang, J.; Gao, R.; Gritsenko, A.; Kingma, D. P .; Poole, B.; Norouzi, M.; Fleet, D. J.; et al. 2022. Imagen video: High definition video generation with diffusion models. arXiv preprint . Ho, J.; Jain, A.; and Abbeel, P . 2020. Denoising diffusion probabilistic models. Advances in neural information pr o- cessing systems , 33: 6840–6851. Kumar , S.; and Byrne, W . 2004. Minimum bayes-risk de- coding for statistical machine translation. T echnical re- port, JOHNS HOPKINS UNIV BAL TIMORE MD CEN- TER FOR LANGU A GE AND SPEECH PR OCESSING (CLSP). Li, X.; Thickstun, J.; Gulrajani, I.; Liang, P . S.; and Hashimoto, T . B. 2022. Diffusion-lm improv es controllable text generation. Advances in neural information pr ocessing systems , 35: 4328–4343. Lin, Z.; Gong, Y .; Shen, Y .; W u, T .; Fan, Z.; Lin, C.; Duan, N.; and Chen, W . 2023. T ext Generation with Diffusion Lan- guage Models: A Pre-training Approach with Continuous Paragraph Denoise. arXi v:2212.11685. Liu, H.; Chen, Z.; Y uan, Y .; Mei, X.; Liu, X.; Mandic, D.; W ang, W .; and Plumbley , M. D. 2023. Audioldm: T ext- to-audio generation with latent diffusion models. arXiv pr eprint arXiv:2301.12503 . Mao, Z.; Zhang, R.; Xin, L.; and Li, M. 2023. Mitigating the missing-fragmentation problem in de nov o peptide sequenc- ing with a two-stage graph-based deep learning model. Na- tur e Machine Intelligence , 5(11): 1250–1260. Moll, J.; and Colombo, R. 2019. T ar get identification and validation in drug discovery . Springer . Qiao, R.; Tran, N. H.; Xin, L.; Chen, X.; Li, M.; Shan, B.; and Ghodsi, A. 2021. Computationally instrument- resolution-independent de novo peptide sequencing for high-resolution devices. Natur e Machine Intelligence , 3(5): 420–425. Rombach, R.; Blattmann, A.; Lorenz, D.; Esser , P .; and Om- mer , B. 2022. High-resolution image synthesis with latent diffusion models. In Proceedings of the IEEE/CVF confer- ence on computer vision and pattern r ecognition , 10684– 10695. Song, Y .; Sohl-Dickstein, J.; Kingma, D. P .; Kumar , A.; Er - mon, S.; and Poole, B. 2020. Score-based generativ e model- ing through stochastic differential equations. arXiv pr eprint arXiv:2011.13456 . Sutske ver , I.; V inyals, O.; and Le, Q. V . 2014. Sequence to sequence learning with neural networks. Advances in neural information pr ocessing systems , 27. T ran, N. H.; Zhang, X.; Xin, L.; Shan, B.; and Li, M. 2017. De novo peptide sequencing by deep learning. Proceedings of the National Academy of Sciences , 114(31): 8247–8252. V aswani, A.; Shazeer , N.; Parmar , N.; Uszkoreit, J.; Jones, L.; Gomez, A. N.; Kaiser, Ł.; and Polosukhin, I. 2017. At- tention is all you need. Advances in neur al information pr o- cessing systems , 30. W atson, J. L.; Juergens, D.; Bennett, N. R.; T rippe, B. L.; Y im, J.; Eisenach, H. E.; Ahern, W .; Borst, A. J.; Ragotte, R. J.; Milles, L. F .; et al. 2023. De novo design of protein structure and function with RFdiffusion. Natur e , 620(7976): 1089–1100. Xia, J.; Chen, S.; Zhou, J.; Xiaojun, S.; Du, W .; Gao, Z.; T an, C.; Hu, B.; Zheng, J.; and Li, S. Z. 2024. Adanov o: T owards robust \ emph { De Novo } peptide sequencing in proteomics against data biases. Advances in Neural Information Pr o- cessing Systems , 37: 1811–1828. Xu, M.; Y u, L.; Song, Y .; Shi, C.; Ermon, S.; and T ang, J. 2022. Geodiff: A geometric diffusion model for molecular conformation generation. arXiv pr eprint arXiv:2203.02923 . Y ang, T .; Ling, T .; Sun, B.; Liang, Z.; Xu, F .; Huang, X.; Xie, L.; He, Y .; Li, L.; He, F .; et al. 2024. Introducing π -HelixNov o for practical large-scale de nov o peptide se- quencing. Briefings in Bioinformatics , 25(2): bbae021. Y ilmaz, M.; Fondrie, W .; Bittremieux, W .; Oh, S.; and No- ble, W . S. 2022. De novo mass spectrometry peptide se- quencing with a transformer model. In International Con- fer ence on Machine Learning , 25514–25522. PMLR. Zhang, L.; Rao, A.; and Agrawala, M. 2023. Adding condi- tional control to text-to-image diffusion models. In Proceed- ings of the IEEE/CVF international confer ence on computer vision , 3836–3847. Zhang, Y .; F onslow , B. R.; Shan, B.; Baek, M.-C.; and Y ates III, J. R. 2013. Protein analysis by shotgun/bottom- up proteomics. Chemical r eviews , 113(4): 2343–2394. Zhou, J.; Chen, S.; Xia, J.; Sizhe Liu, S.; Ling, T .; Du, W .; Liu, Y .; Y in, J.; and Li, S. Z. 2024. Novobench: Bench- marking deep learning-based \ emph { De Nov o } sequencing methods in proteomics. Advances in Neural Information Pr ocessing Systems , 37: 104776–104791.
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment