베타이항 가우시안 프로세스로 시계열 고처리량 시퀀싱 데이터 분석
** 본 논문은 고처리량 시퀀싱(HTS) 기반 실험 진화에서 시간에 따라 변하는 대립유전자 빈도를 분석하기 위해, 시퀀싱 깊이의 불확실성을 베타‑이항 모델로 추정하고 이를 가우시안 프로세스(GP)와 결합한 베타‑이항 가우시안 프로세스(BBGP) 검정을 제안한다. 시뮬레이션과 실제 초파리 데이터에서 기존의 Cochran‑Mantel‑Haenszel(CMH) 검정보다 높은 평균 정밀도를 보이며, 실험 설계 선택에 대한 탐색도 제공한다. *…
저자: H, e Topa, Agnes Jonas
**
본 논문은 고처리량 시퀀싱(HTS) 기술이 발달함에 따라, 단일 시점이 아닌 여러 시점에 걸친 유전체 데이터를 활용해 동적인 생물학적 현상을 분석하려는 요구가 증가하고 있음을 출발점으로 한다. 특히 실험 진화 분야에서는 대립유전자 빈도(allele frequency, AF)의 시간적 변화를 추적함으로써 선택 압력이나 유전적 부동(drift)을 정량화하고자 한다. 기존의 분석 방법은 주로 초기와 최종 시점 사이의 차이를 Fisher’s exact test, Cochran‑Mantel‑Haenszel(CMH) 등으로 검정했으며, 이는 중간 시점 데이터와 복제 간 변동성을 충분히 활용하지 못한다는 한계가 있었다.
이를 극복하기 위해 저자들은 두 가지 통계적 모델을 결합한 베타‑이항 가우시안 프로세스(BBGP) 프레임워크를 제안한다. 첫 단계는 베타‑이항 모델을 이용해 각 시점·복제마다 측정된 대립유전자 카운트(y_ij)와 전체 깊이(n_ij)로부터 대립유전자 비율(p_ij)의 사후 평균(m_ij)과 분산(s^2_ij)을 추정한다. 베타 사전(α=β=1)을 사용함으로써 사후 분포는 베타(α*ij,β*ij) 형태가 되며, 이는 관측 깊이가 낮을수록 큰 분산을 제공한다. 이 단계는 HTS 데이터 특유의 불확실성을 정량화하는 핵심 전처리 과정이다.
두 번째 단계에서는 시간에 따른 AF 변화를 비모수적으로 모델링하기 위해 가우시안 프로세스(GP)를 도입한다. GP는 임의의 시점 집합에 대해 다변량 정규분포를 가정하며, 여기서는 평균을 0으로 두고(관측값 평균을 빼는 전처리) 제곱 지수 커널(K_SE)로 공분산을 정의한다. K_SE는 길이 척도(l)와 신호 분산(σ_f^2) 두 파라미터를 갖는다. 관측 노이즈는 두 부분으로 구성된다. 첫째, 전통적인 백색 노이즈 Σ_W=σ_n^2 I; 둘째, 베타‑이항 단계에서 얻은 고정 분산 Σ_FBB=diag(s^2_ij) 로, 후자는 각 관측치마다 사전 추정된 불확실성을 그대로 반영한다. 이렇게 하면 깊이가 낮은 시점에서는 큰 노이즈가, 깊이가 높은 시점에서는 작은 노이즈가 모델에 투입되어, 실제 데이터의 이질성을 정확히 반영한다.
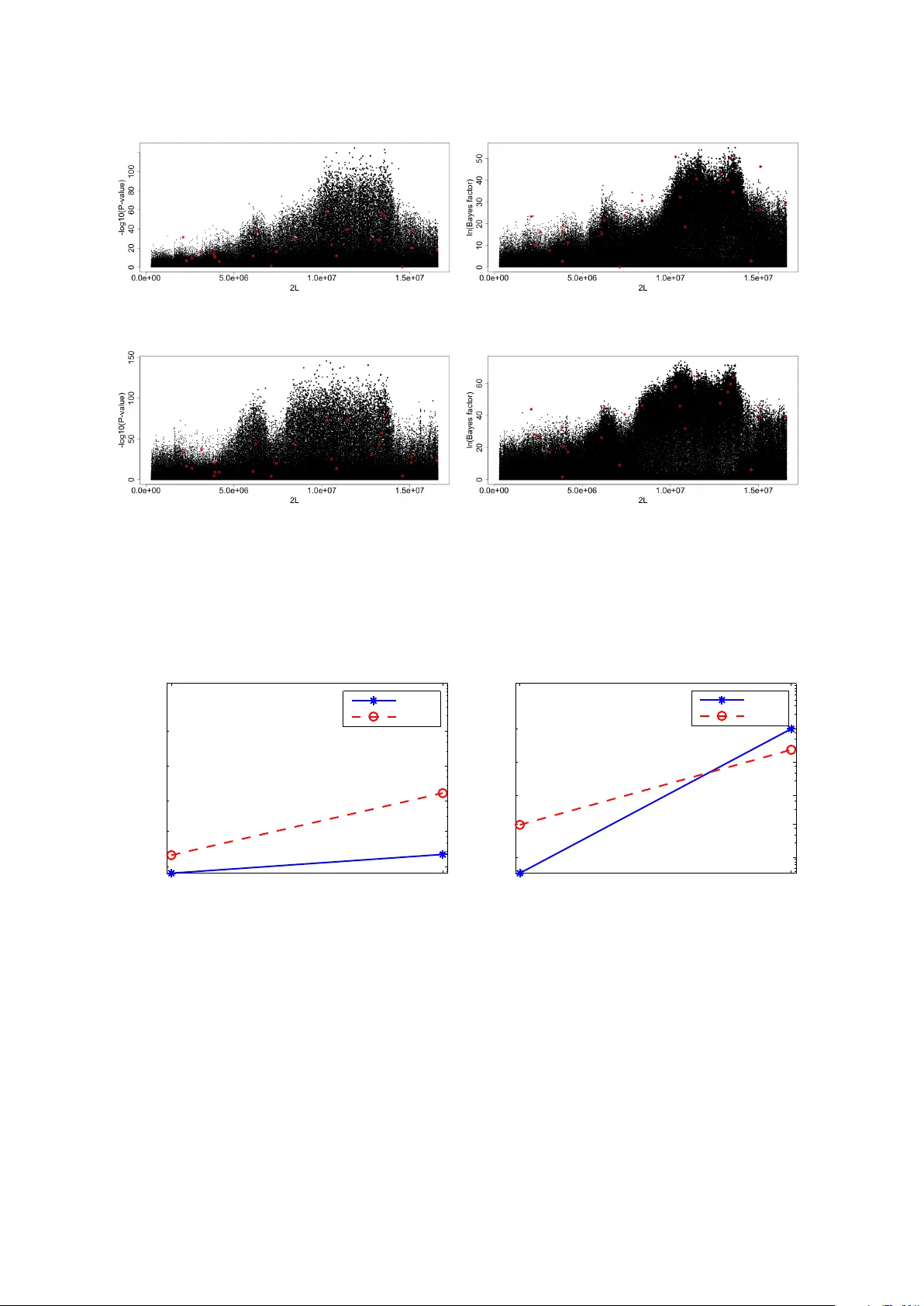
모델 적합은 두 개의 대립 모델을 비교함으로써 수행된다. (1) 시간‑의존 모델: m_ij = f_i(t_j) + ε_ij, 여기서 f_i(t) ~ GP(0, K_SE)이며 ε_ij ~ N(0, σ_n^2 + s^2_ij). (2) 시간‑비의존 모델: m_ij = μ_i + ε_ij, 즉 GP 항이 없고 관측값이 일정 평균 주위에 흩어진다고 가정한다. 두 모델 각각에 대해 로그 주변우도(log‑marginal‑likelihood)를 최대화하여 파라미터 θ̂_1, θ̂_2를 얻는다. 이후 베이즈 팩터 BF_i = p(m_i|θ̂_1, time‑dep) / p(m_i|θ̂_2, time‑indep) 를 계산하고, BF가 큰 SNP를 선택 후보로 삼는다. BF는 모델이 시간 의존성을 얼마나 잘 설명하는지를 정량화하는 지표이며, 기존 p‑값 기반 검정보다 연속적인 증거 강도를 제공한다.
알고리즘 구현은 R 패키지 “BBGP”로 제공되며, gptk 패키지를 활용해 스케일드 컨쥐게이트 그라디언트 방법으로 최적화를 수행한다. 초기값은 파라미터 공간을 격자 탐색한 뒤 가장 높은 우도를 보인 지점으로 설정하고, 길이 척도 l에 대한 하한을 관측 시점 간 최소 간격으로 제한해 과적합을 방지한다.
성능 평가는 두 차원에서 진행된다. 첫째, 실험 진화 시뮬레이션을 통해 BBGP와 CMH 검정을 비교한다. 시뮬레이션은 유효 개체수(Ne) 10^2–10^3, 세대 수 5–10, 복제 3–5, 다양한 시퀀싱 깊이(10–60) 등을 변형시켜 실제 실험 조건을 재현한다. 결과는 평균 정밀도(average precision)와 ROC 곡선에서 BBGP가 특히 낮은 깊이와 짧은 시계열 상황에서 CMH보다 현저히 높은 성능을 보였으며, 베이즈 팩터 기반 순위가 실제 선택된 SNP를 더 잘 재현함을 확인했다. 둘째, 실제 초파리(Drosophila melanogaster) 온도 적응 실험 데이터에 적용하였다. BBGP는 CMH보다 더 많은 알려진 적응 관련 유전자와 겹치는 SNP를 식별했으며, 시계열 전체를 활용함으로써 복제 간 일관된 변화를 포착했다. 또한 실험 설계 변수(복제 수, 시점 수, 깊이)의 민감도 분석을 통해, 복제와 시점이 충분히 많을 경우 BBGP의 이점이 크게 확대된다는 점을 제시한다.
결론적으로, 베타‑이항으로 추정한 관측 불확실성을 GP에 직접 통합함으로써, 짧고 불규칙적인 HTS 시계열 데이터에서도 과적합 없이 안정적인 변이 탐지가 가능해졌다. BBGP는 기존의 pairwise 검정이 놓치기 쉬운 중간 시점 정보와 복제 간 변동성을 동시에 활용함으로써, 실험 진화뿐 아니라 시간에 따라 변하는 메틸화, 전사체, 메타게놈 데이터 등 다양한 HTS 기반 시계열 연구에 적용 가능하다. 제공된 오픈소스 R 구현은 사용자가 손쉽게 시뮬레이션, 모델 적합, 베이즈 팩터 계산까지 전 과정을 수행하도록 설계되어, 향후 HTS 시계열 분석의 표준 도구로 자리매김할 잠재력을 가진다.
**
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기