암 유전자 발현 차이를 찾는 새로운 통계법 MOST
본 논문은 암 조직에서 소수의 샘플만 발현이 증가하는 유전자를 탐지하기 위해 “Maximum Ordered Subset t‑statistics”(MOST)를 제안한다. 기존의 t‑검정, COPA, OS, ORT와 비교한 시뮬레이션 및 실제 유방암 마이크로어레이 데이터 분석 결과, MOST는 특히 활성화된 샘플 수가 알려지지 않았을 때 전반적으로 높은 검출력을 보이며, 일부 상황에서는 기존 방법보다 유의미하게 우수함을 확인하였다.
저자: ** - **Heng Lian** (주 저자) *(논문 원문에 다른 공동 저자 정보가 명시되지 않아, 제공된 저자명만 기재)* --- **
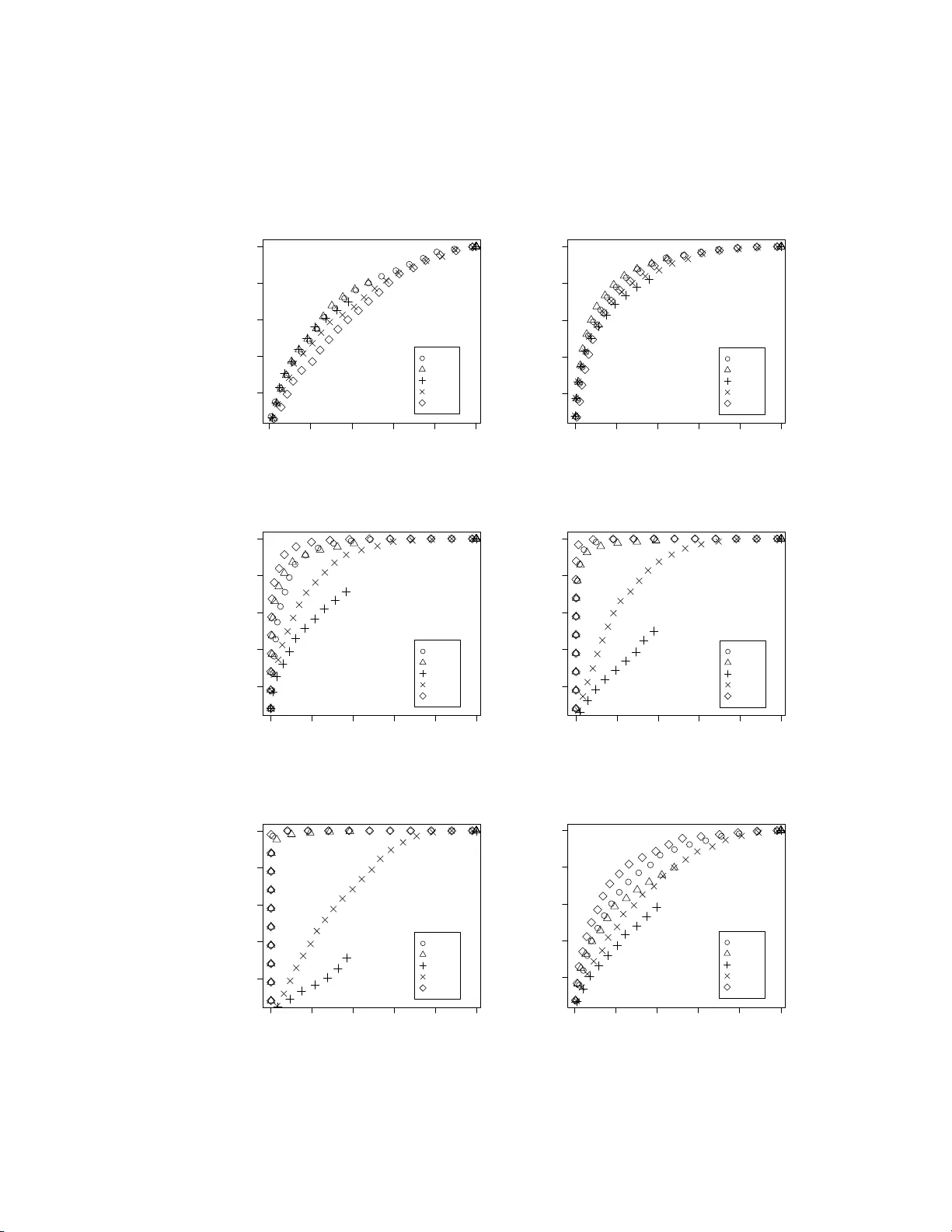
본 논문은 암 연구에서 흔히 나타나는 “희소 활성화” 현상을 통계적으로 탐지하기 위한 새로운 방법론, 즉 Maximum Ordered Subset t‑statistics (MOST)를 제안한다. 전통적인 두 집단 t‑검정은 전체 암군이 평균적으로 정상군보다 높은 발현을 보인다고 가정한다. 그러나 실제 암 데이터에서는 특정 유전자가 소수의 종양 샘플에서만 현저히 과발현되는 경우가 많으며, 이러한 경우 t‑검정은 검출력이 크게 저하된다. 이를 보완하기 위해 기존에 제안된 방법으로는 Tomlins et al.이 제시한 COPA, Tibshirani‑Hastie의 Outlier Sum (OS), Wu의 Outlier Robust t‑statistics (ORT) 등이 있다. 이들 방법은 각각 특정 백분위수 혹은 이상치 집합을 정의하고, 그 값을 이용해 통계량을 구성한다. 그러나 이상치 정의에 사용되는 임계값이 주관적이며, 활성화된 샘플 수 k가 사전에 알려지지 않은 경우 최적의 k를 선택하기 어렵다는 공통된 한계가 있다.
MOST는 이러한 문제를 근본적으로 해결한다. 먼저 각 유전자 i에 대해 암 샘플의 발현값을 내림차순으로 정렬한다(y_{i1}≥y_{i2}≥…≥y_{im}). 그런 다음 k=1,…,m에 대해 상위 k개의 값과 정상군 중앙값(또는 평균) 차이의 합을 구한다. 이때 차이를 표준화하기 위해 MAD(중앙값 절대 편차)로 나누고, 표준 정규분포에서 얻은 순위통계량의 기대값 μ_k와 분산 σ_k^2를 이용해 정규화한다. 즉, M_{ik}= (∑_{j=1}^k (y_{ij}−med_x)/MAD − μ_k)/σ_k 로 정의한다. 이렇게 하면 k가 달라져도 M_{ik}는 동일한 표준 정규분포를 따르므로 직접 비교가 가능해진다. 최종 통계량 M_i = max_{k} M_{ik}는 “가장 강력한” 이상치 집합을 자동으로 선택한다는 의미이며, k를 별도로 추정할 필요가 없다는 장점을 가진다.
논문에서는 이 방법의 성능을 평가하기 위해 두 가지 접근을 사용하였다. 첫째, 시뮬레이션 실험에서는 정상 샘플과 암 샘플을 각각 n=m=20으로 설정하고, 다양한 k (활성화된 샘플 수)와 μ (발현 차이) 값을 조합하였다. 각 조건에서 1,000개의 차등 발현 유전자와 1,000개의 비차등 유전자를 생성하고, ROC 곡선을 통해 검출력을 비교하였다. 결과는 다음과 같다. μ=2, k가 작을 때는 모든 방법이 비슷한 성능을 보였지만, k가 증가함에 따라 전통적인 t‑검정이 우세해졌다. μ=1, 중간~큰 k에서는 MOST가 t‑검정보다 약간 뒤처지지만 다른 방법들보다 확연히 앞섰다. μ=4, k가 매우 작을 때는 MOST가 ORT, COPA, t‑검정을 모두 능가했으며, OS와 거의 동등한 수준을 유지했다. 이러한 결과는 MOST가 “극소수 샘플만 활성화된 경우”와 “중간 정도의 활성화가 존재하는 경우” 모두에서 안정적인 검출력을 제공함을 시사한다.
둘째, 실제 데이터 적용에서는 West et al. (2001)의 유방암 마이크로어레이 데이터를 사용하였다. 이 데이터는 7,129개의 유전자와 49개의 종양 샘플(양성·음성 각각 24, 25)로 구성되어 있다. 로그 변환 및 정규화 후 MOST와 전통적인 t‑검정을 각각 SAM (Significance Analysis of Microarrays) 기반 FDR 추정과 결합하였다. FDR 대 호출된 유전자 수 그래프에서 MOST는 t‑검정보다 약간 낮은 FDR을 보였으나 차이는 통계적으로 유의미하지 않았다. 이는 실제 데이터에서 두 방법이 비슷한 수준의 신호‑노이즈 비율을 갖고 있음을 의미한다.
논문은 몇 가지 한계를 명시한다. 첫째, null 분포를 표준 정규로 가정했지만 실제 마이크로어레이 데이터는 비정규성을 보일 수 있다. 둘째, 다중 검정 보정에 대한 상세한 논의가 부족하고, FDR 제어를 위한 다양한 방법과의 비교가 이루어지지 않았다. 셋째, MOST의 계산 복잡도는 O(p·m·log m) 정도로, 대규모 RNA‑seq 데이터(수만 유전자, 수백 샘플)에서는 효율적인 구현이 필요하다. 마지막으로, 활성화된 샘플 수 k를 추정하는 부가적인 해석이 요구되는데, 논문에서는 M_i의 argmax를 “추정값”으로 제시했지만, 그 정확도와 신뢰구간에 대한 검증이 부족하다.
결론적으로, MOST는 기존 방법들의 임계값 선택 문제를 통계적으로 정규화된 프레임워크 안에서 해결하고, 활성화된 샘플 수가 불확실한 상황에서도 강인한 검출력을 제공한다는 점에서 의미가 크다. 다만 실제 데이터의 복잡성을 충분히 반영한 추가 검증과 구현 최적화가 뒤따라야 할 것이다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기