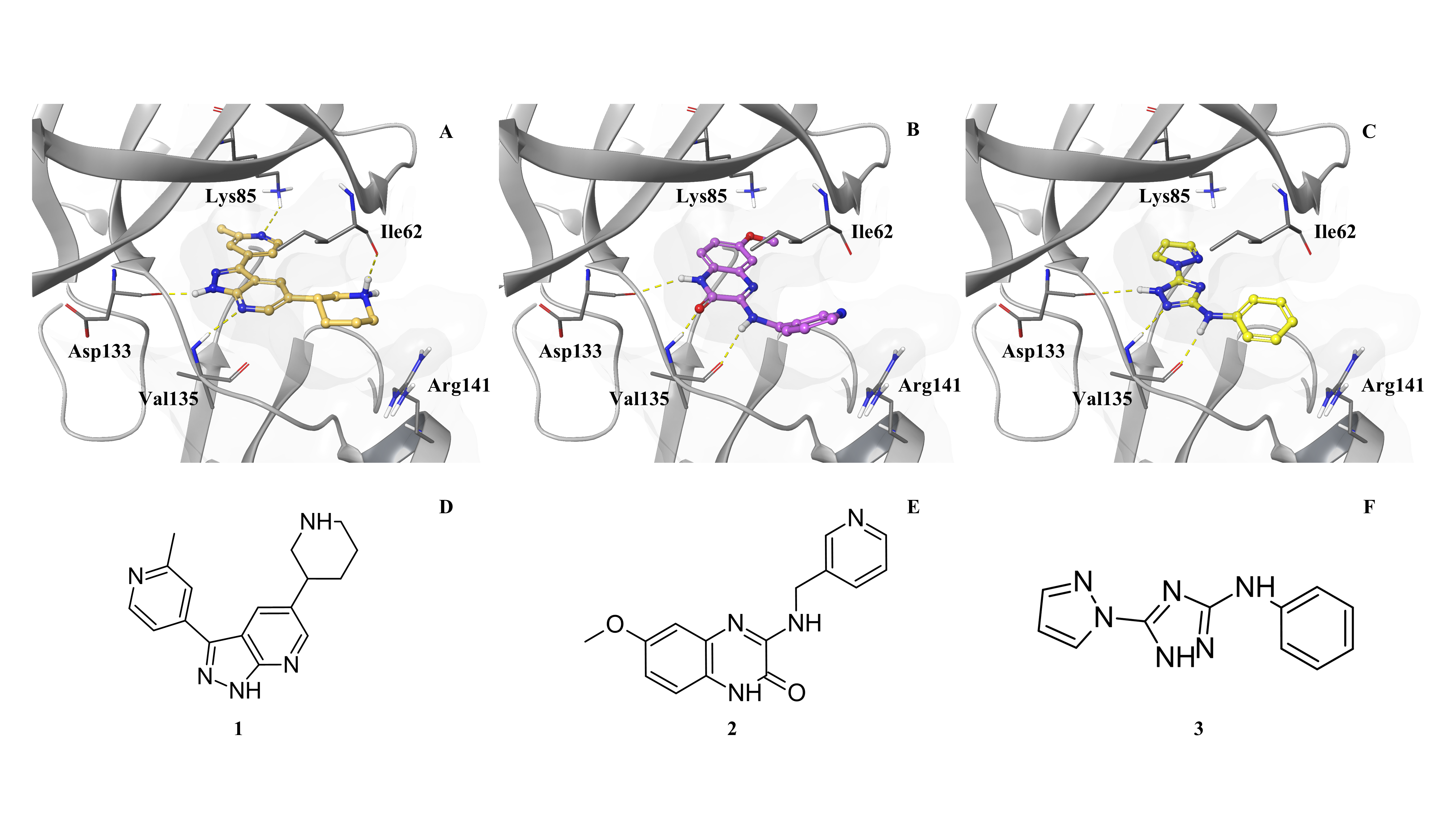
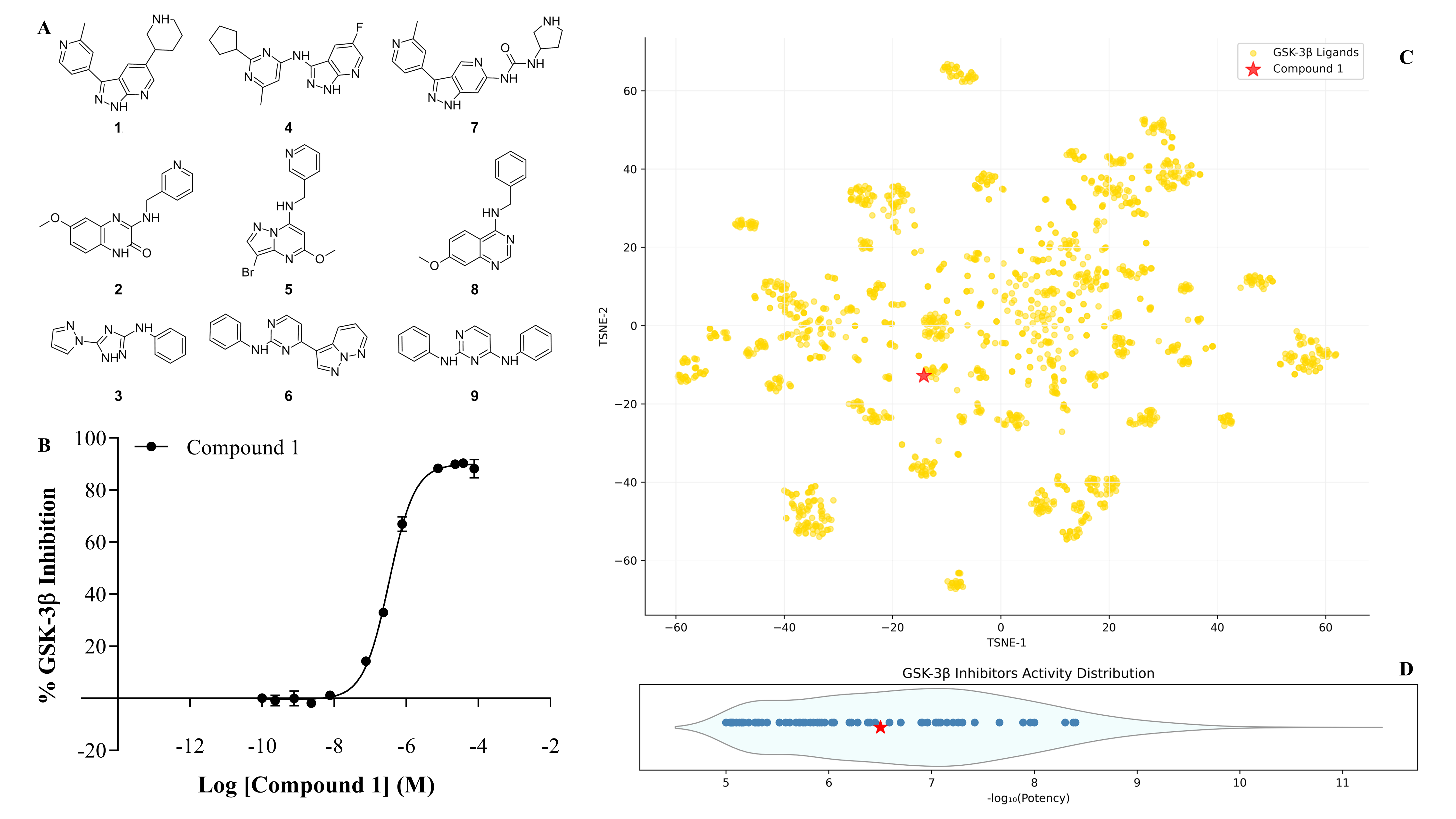
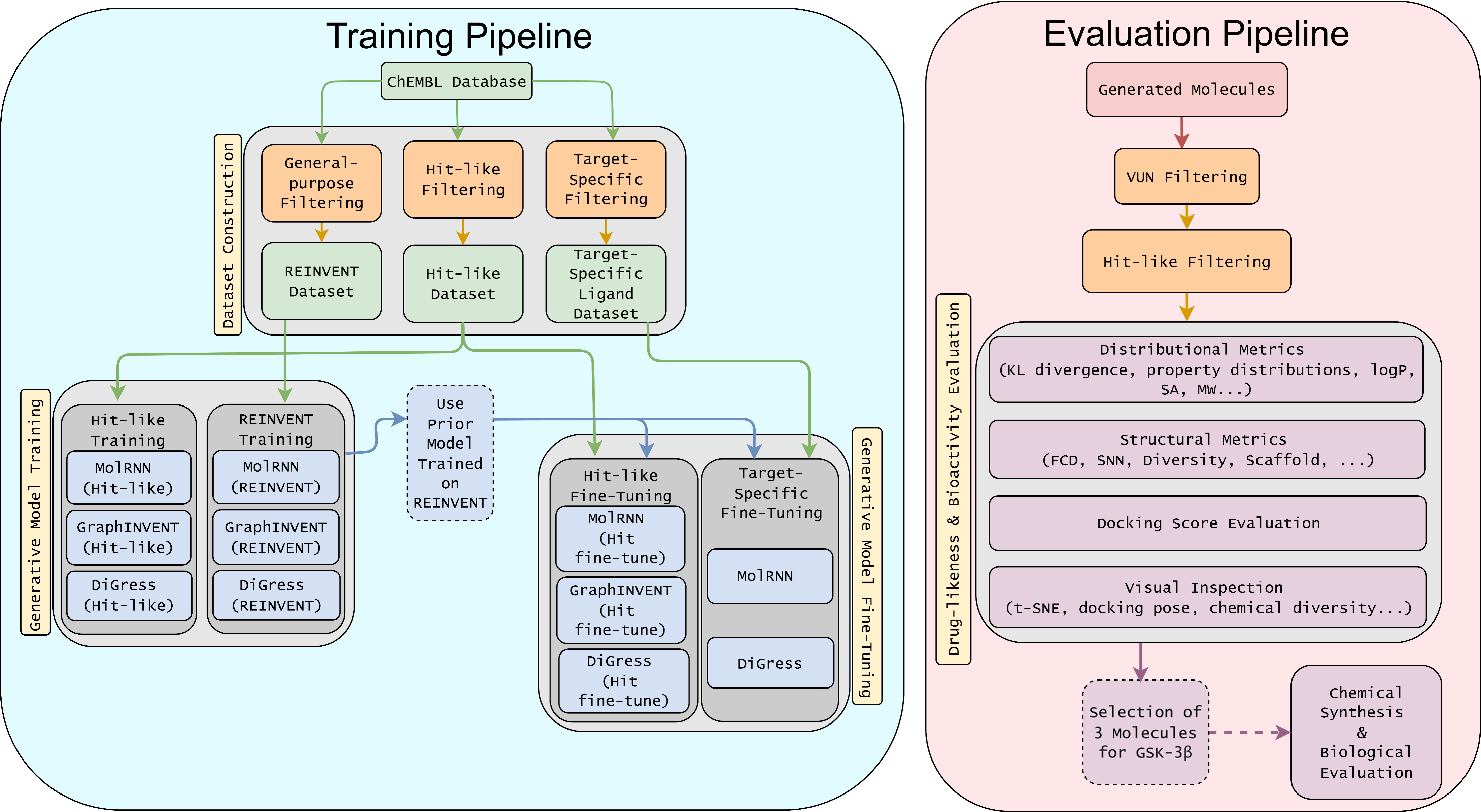
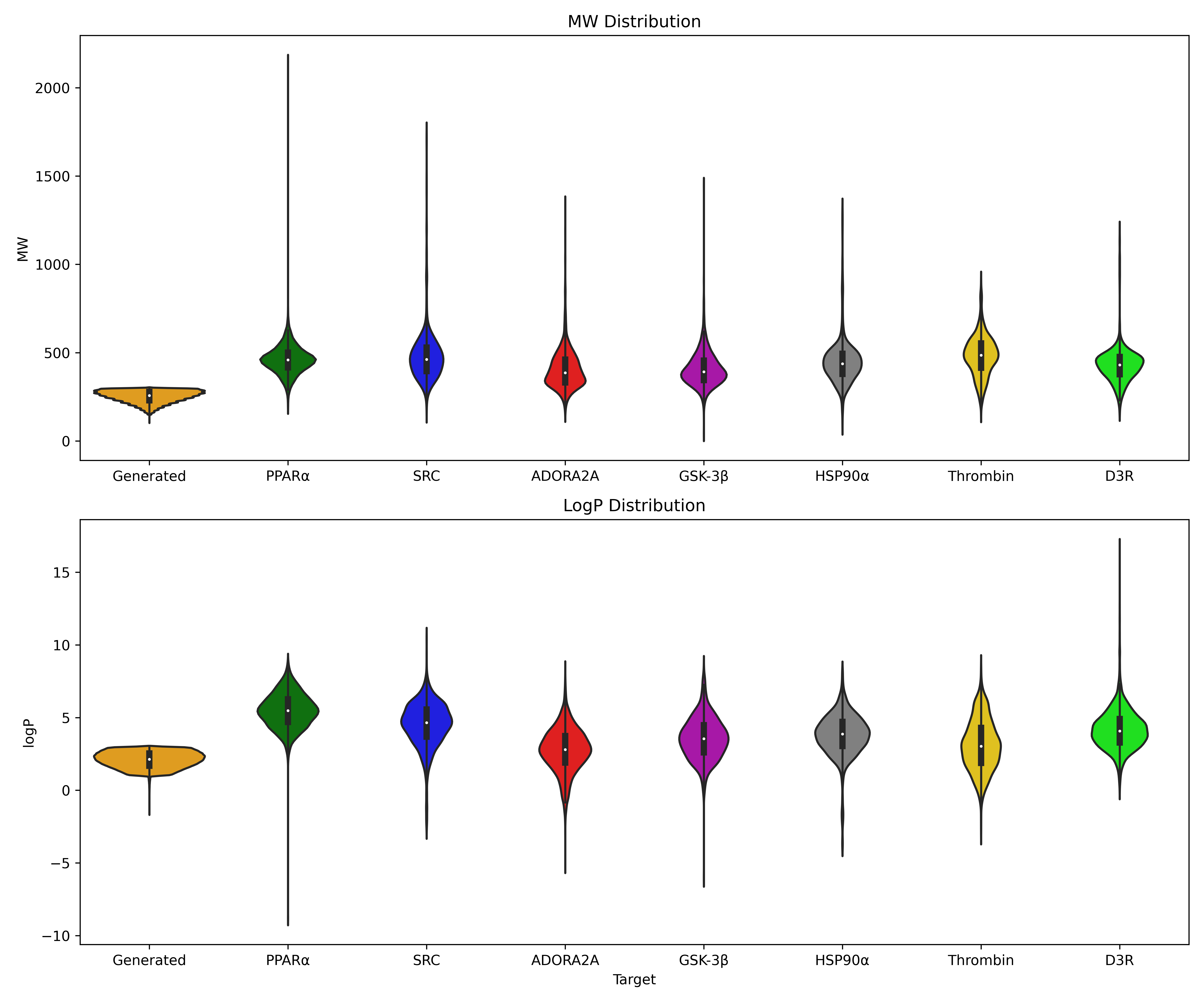
Title: From In Silico to In Vitro: Evaluating Molecule Generative Models for Hit Generation
ArXiv ID: 2512.22031
Date: 2025-12-26
Authors: Nagham Osman, Vittorio Lembo, Giovanni Bottegoni, Laura Toni
📝 Abstract
Hit identification is a critical yet resource-intensive step in the drug discovery pipeline, traditionally relying on high-throughput screening of large compound libraries. Despite advancements in virtual screening, these methods remain time-consuming and costly. Recent progress in deep learning has enabled the development of generative models capable of learning complex molecular representations and generating novel compounds de novo. However, using ML to replace the entire drug-discovery pipeline is highly challenging. In this work, we rather investigate whether generative models can replace one step of the pipeline: hit-like molecule generation. To the best of our knowledge, this is the first study to explicitly frame hit-like molecule generation as a standalone task and empirically test whether generative models can directly support this stage of the drug discovery pipeline. Specifically, we investigate if such models can be trained to generate hit-like molecules, enabling direct incorporation into, or even substitution of, traditional hit identification workflows. We propose an evaluation framework tailored to this task, integrating physicochemical, structural, and bioactivity-related criteria within a multi-stage filtering pipeline that defines the hit-like chemical space. Two autoregressive and one diffusion-based generative models were benchmarked across various datasets and training settings, with outputs assessed using standard metrics and target-specific docking scores. Our results show that these models can generate valid, diverse, and biologically relevant compounds across multiple targets, with a few selected GSK-3$β$ hits synthesized and confirmed active in vitro. We also identify key limitations in current evaluation metrics and available training data.
💡 Deep Analysis
📄 Full Content
From In Silico to In Vitro: Evaluating Molecule
Generative Models for Hit Generation
Nagham Osman
University College London
London, UK
nagham.osman.21@ucl.ac.uk
Vittorio Lembo
University of Urbino Carlo Bo
Urbino, Italy
Giovanni Bottegoni
University of Urbino Carlo Bo
Urbino, Italy
Laura Toni
University College London
London, UK
Abstract
Hit identification is a critical yet resource-intensive step in the drug discovery
pipeline, traditionally relying on high-throughput screening of large compound
libraries. Despite advancements in virtual screening, these methods remain time-
consuming and costly. Recent progress in deep learning has enabled the develop-
ment of generative models capable of learning complex molecular representations
and generating novel compounds de novo. However, using ML to replace the entire
drug-discovery pipeline is highly challenging. In this work, we rather investigate
whether generative models can replace one step of the pipeline: hit-like molecule
generation. To the best of our knowledge, this is the first study to explicitly frame
hit-like molecule generation as a standalone task and empirically test whether
generative models can directly support this stage of the drug discovery pipeline.
Specifically, we investigate if such models can be trained to generate hit-like
molecules, enabling direct incorporation into, or even substitution of, traditional
hit identification workflows. We propose an evaluation framework tailored to this
task, integrating physicochemical, structural, and bioactivity-related criteria within
a multi-stage filtering pipeline that defines the hit-like chemical space. Two au-
toregressive and one diffusion-based generative models were benchmarked across
various datasets and training settings, with outputs assessed using standard metrics
and target-specific docking scores. Our results show that these models can generate
valid, diverse, and biologically relevant compounds across multiple targets, with
a few selected GSK-3β hits synthesized and confirmed active in vitro. We also
identify key limitations in current evaluation metrics and available training data.
1
Introduction
Traditionally, drug discovery begins with target validation, followed by hit identification which is
the first stage introducing chemical matter and novelty. A hit is a small molecule with reproducible
activity, acceptable synthetic accessibility, and physicochemical properties [Goodnow, 2006]. Identi-
fying high-quality hits is critical, as it initiates the hit-to-lead process to improve potency, selectivity,
and pharmacokinetic properties. To guide this process, medicinal chemists often rely on heuristics
such as Lipinski’s Rule of Five, which defines drug-likeness based on molecular weight, lipophilicity,
hydrogen-bonding capacity, and related efficiency metrics that balance potency with physicochemical
properties. Despite its importance, hit identification remains a resource-intensive task. Various
experimental strategies have been developed, including high-throughput screening (HTS) of libraries
of commercially available compounds, in-house collections, or natural products, aimed at identifying
NeurIPS 2025 AI for Science Workshop.
arXiv:2512.22031v1 [cs.LG] 26 Dec 2025
biologically active hits. However, the main bottleneck lies in the fact that these approaches are
both time-consuming (months to years) [Paul et al., 2010] and financially demanding [Hughes et al.,
2011]. To partially overcome these limitations, computational methods such as structure-based virtual
screening have been adopted to efficiently prioritize promising molecules. Nevertheless, even with
these tools, identifying high-quality hits from vast chemical libraries continues to represent a major
bottleneck in drug discovery, as these methods also entail considerable time and resource demands.
In recent years, deep learning (DL) has gained interest in molecular design due to its potential to learn
complex structures and property relationships from large chemical datasets. To address persistent
challenges in early drug discovery for de novo molecule generation, research has increasingly explored
generative models that are trained either to replicate the distribution of known compounds or to
optimize specific constraints, such as molecular properties or binding affinity. Common strategies
include one-shot generation [De Cao and Kipf, 2018, Vignac et al., 2023, Zang and Wang, 2020],
sequential atom- or bond-level construction [Gebauer et al., 2022, Segler et al., 2018, Zhou et al.,
2019], and fragment-based assembly [Podda et al., 2020, Seo et al., 2021, Jin et al., 2020, Gupta
et al., 2018]. However, prior research on generative models has predominantly focused on generating
molecules in general, without considering how such models perform when applied to a specific
step in the drug discovery process. This work investigates their applicability to a more specific
and demanding task: generating hit molecules. It reframes t