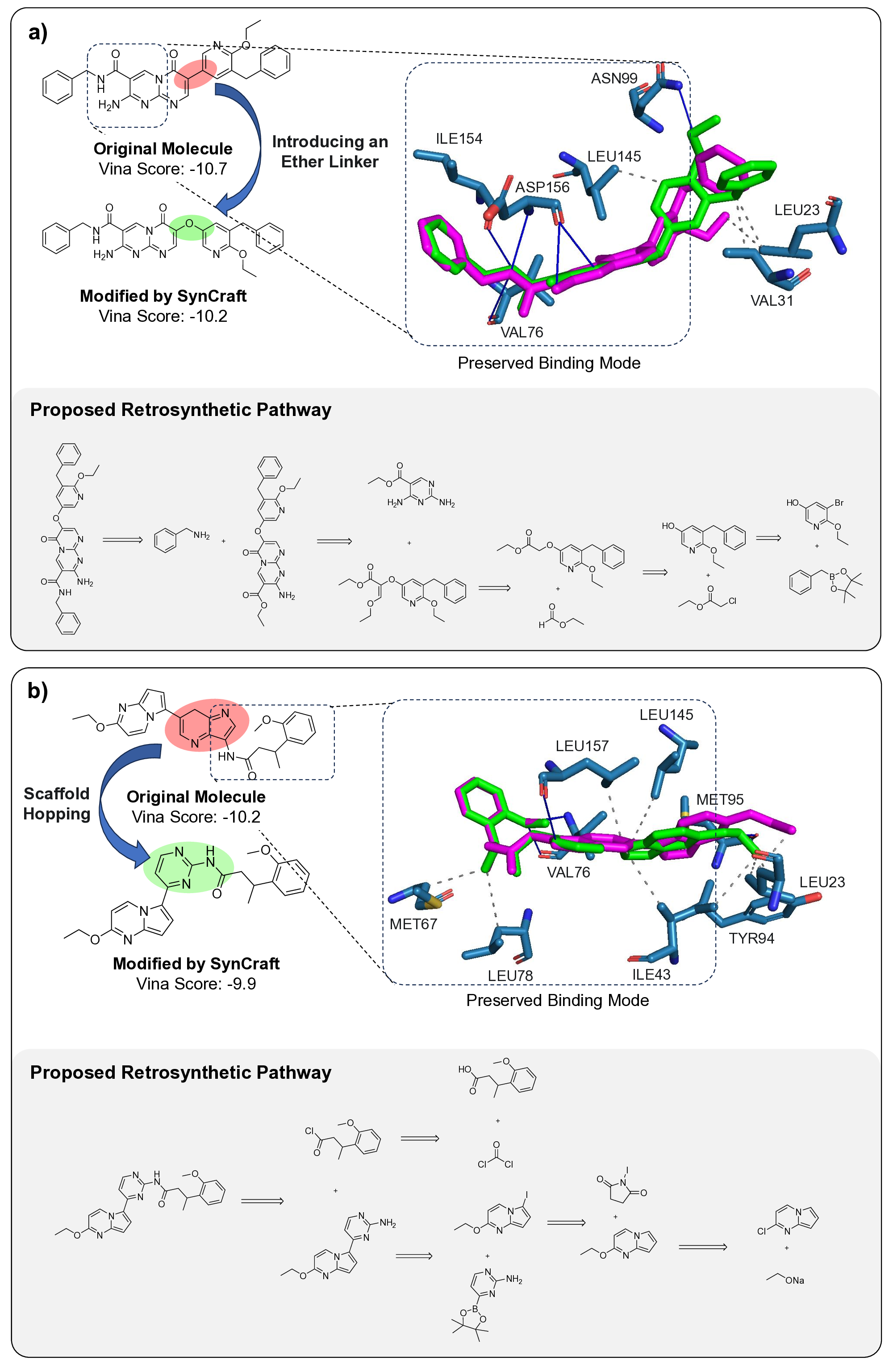
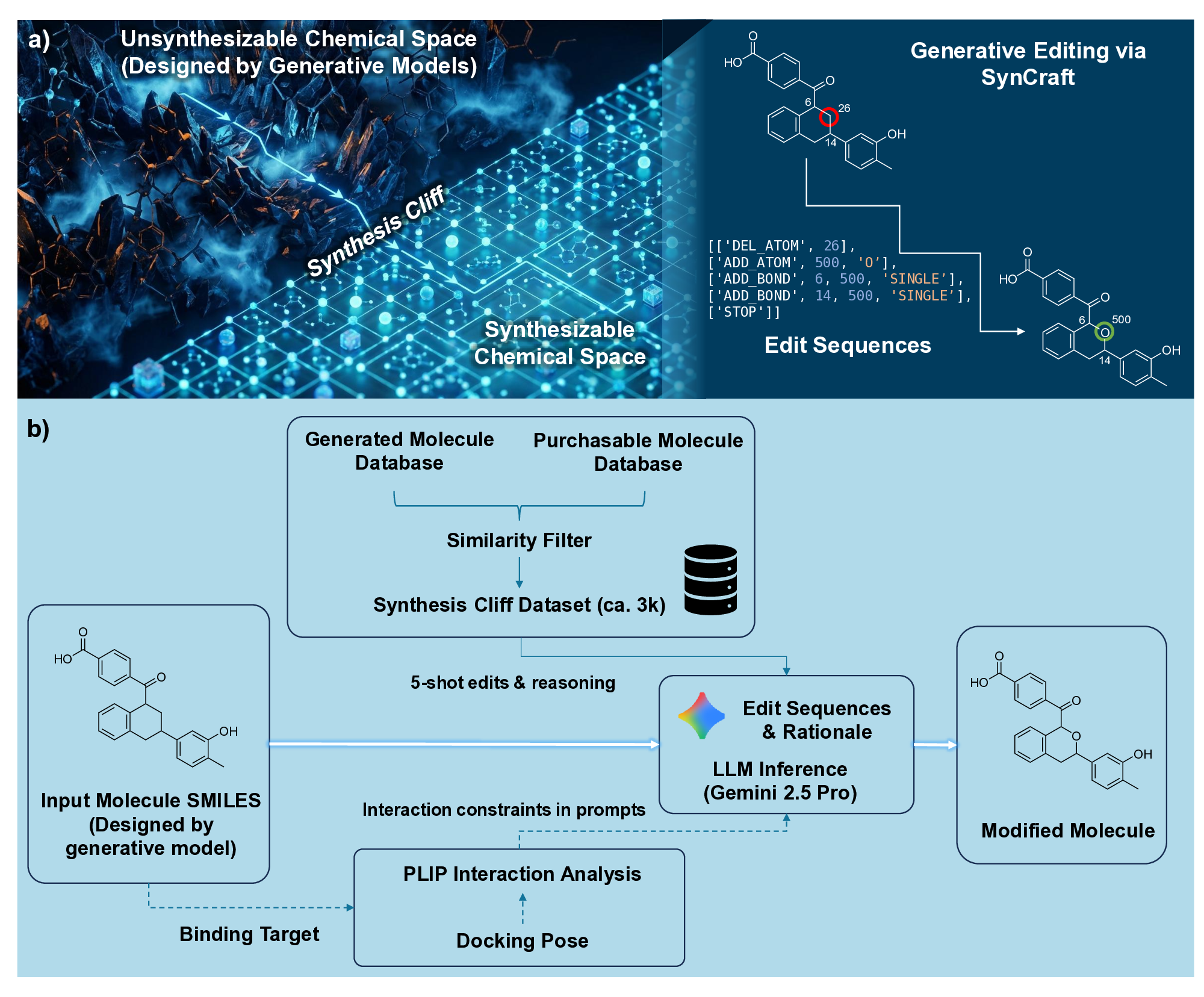
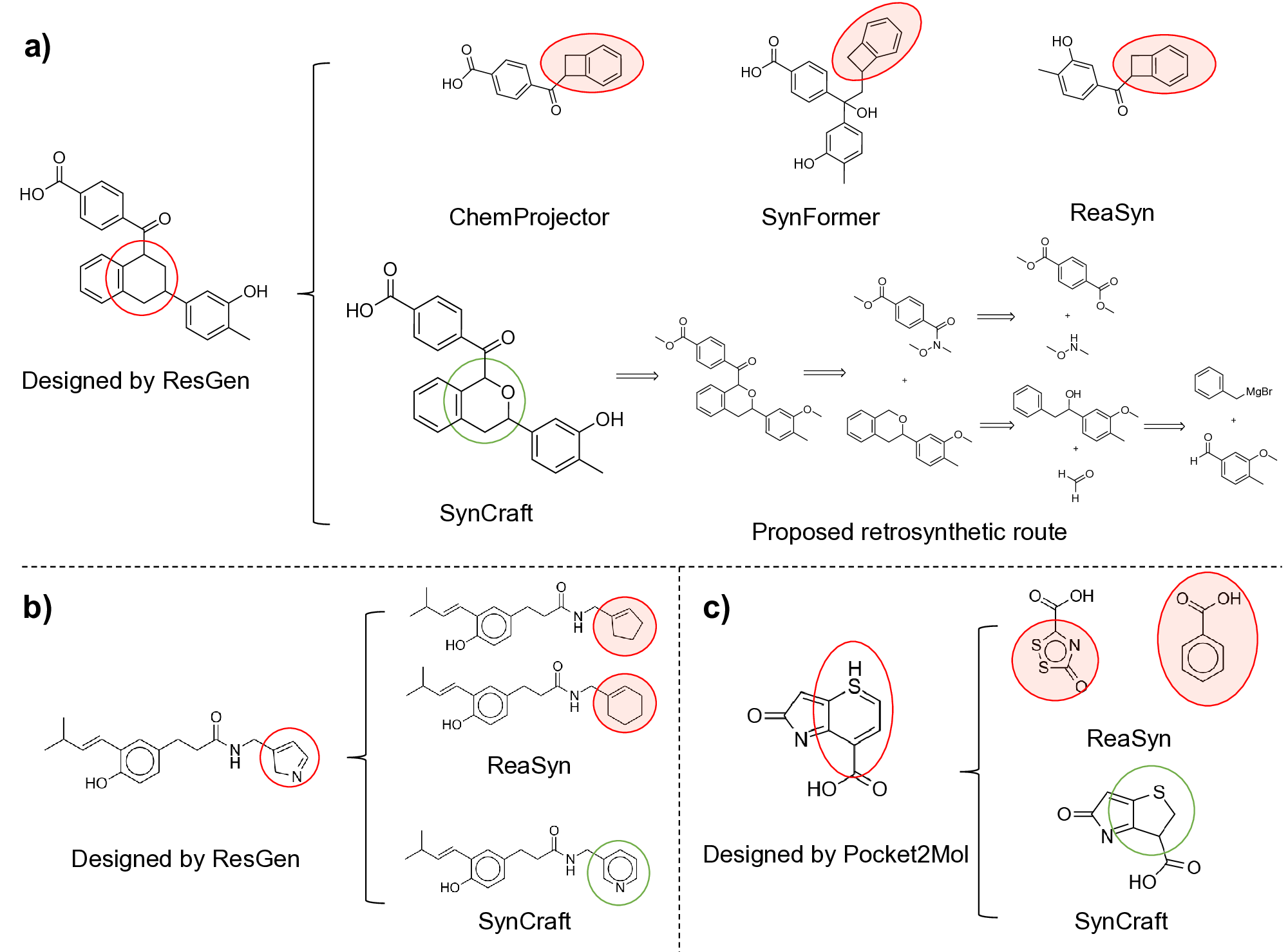
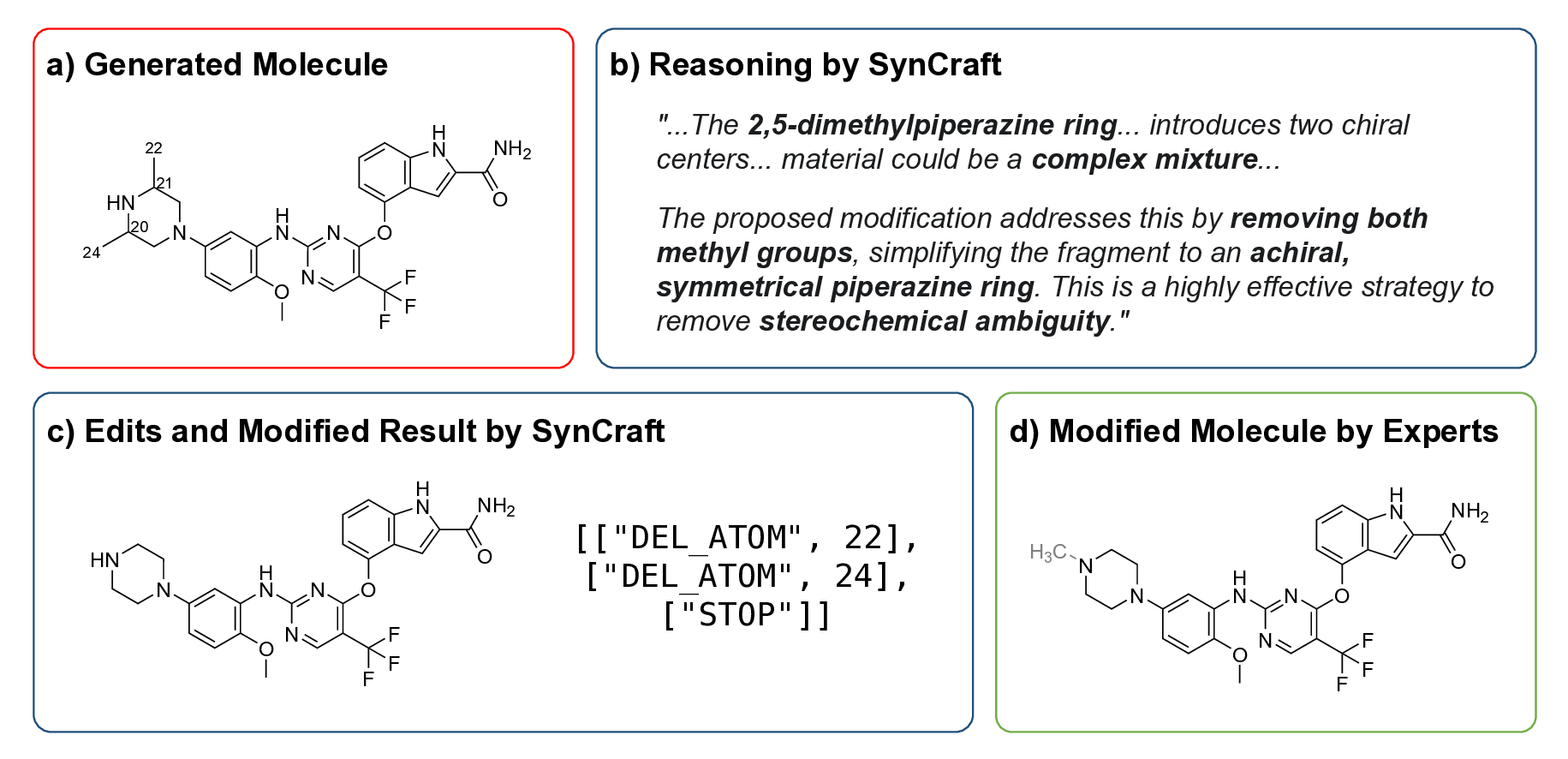
Generative artificial intelligence has revolutionized the exploration of chemical space, yet a critical bottleneck remains that a substantial fraction of generated molecules is synthetically inaccessible. Current solutions, such as post-hoc filtering or projection-based methods, often compromise structural novelty or disrupt key pharmacophores by forcing molecules into pre-defined synthetic templates. Herein, we introduce SynCraft, a reasoning-based framework that reframes synthesizability optimization not as a sequence translation task, but as a precise structural editing problem. Leveraging the emergent reasoning capabilities of Large Language Models, SynCraft navigates the "synthesis cliff" where minimal structural modifications yield significant gains in synthetic feasibility. By predicting executable sequences of atom-level edits rather than generating SMILES strings directly, SynCraft circumvents the syntactic fragility of LLMs while harnessing their chemical intuition. Extensive benchmarks demonstrate that SynCraft outperforms state-of-the-art baselines in generating synthesizable analogs with high structural fidelity. Furthermore, through interaction-aware prompting, SynCraft successfully replicates expert medicinal chemistry intuition in editing PLK1 inhibitors and rescuing high-scoring but previously discarded RIPK1 candidates in previous molecular generation literatures.
The discovery and development of new therapeutics is always a lengthy, costly, and high-risk endeavor. 1 In recent years, artificial intelligence-driven drug discovery (AIDD) has emerged as a transformative paradigm to accelerate this process. 2,3 One of the key techniques in AIDD is generative models, which have demonstrated a remarkable capacity to explore the vastness of chemical space. 4,5 The primary objective of these models is often narrowly focused on optimizing a specific property, most notably the predicted binding affinity to a protein target. In their pursuit of maximizing this objective function, however, such models frequently operate with an incomplete understanding of chemical principles, generating structures that, while computationally optimal, exhibit poor stability or contain strained, synthetically infeasible motifs. This intense focus on a single endpoint, often at the expense of holistic chemical reasonableness, has created a critical bottleneck: a significant portion of the most promising generated hits are practically impossible to synthesize, impeding their translation into tangible laboratory assets. [6][7][8][9][10] To bridge this gap between virtual design and real-world synthesis, several strategies have been proposed. [11][12][13] One approach involves post-hoc filtering, where generated candidates are screened using retrosynthesis software, a process that ensures synthesizability but often discards a vast number of potentially valuable molecules 8,14,15 Alternatively, other methods constrain the generation process a priori by constructing molecules exclusively from a finite library of building blocks and reaction templates, [16][17][18][19] or by projecting a generated molecule onto its nearest analog within such a pre-defined synthesizable space. [20][21][22][23][24] While effective to an extent, these methods impose a fundamental trade-off, which sacrifices the very exploratory power that makes generative models attractive. The explorable chemical space is drastically curtailed, and promising scaffolds that lie just outside these rigid boundaries are lost.
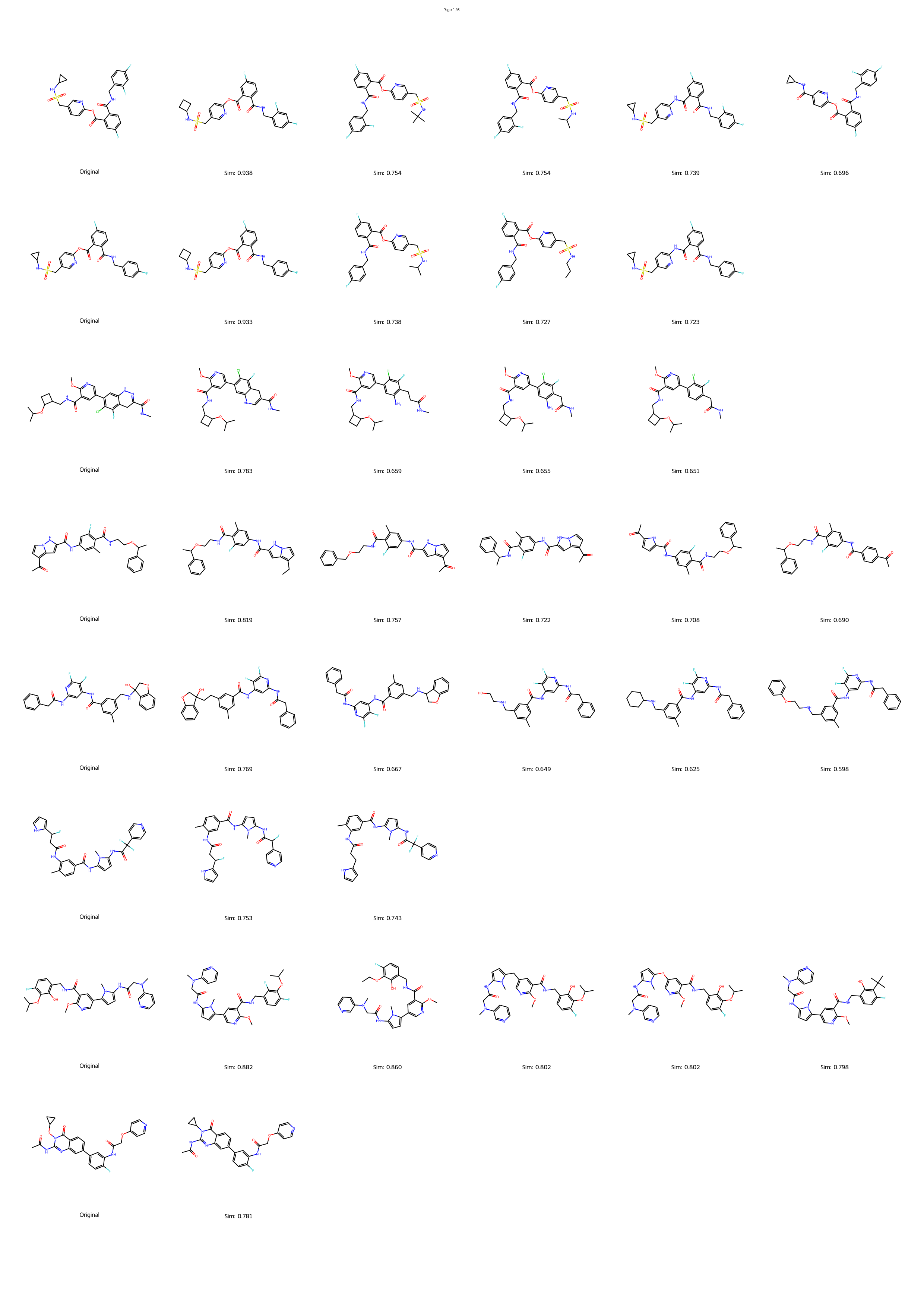
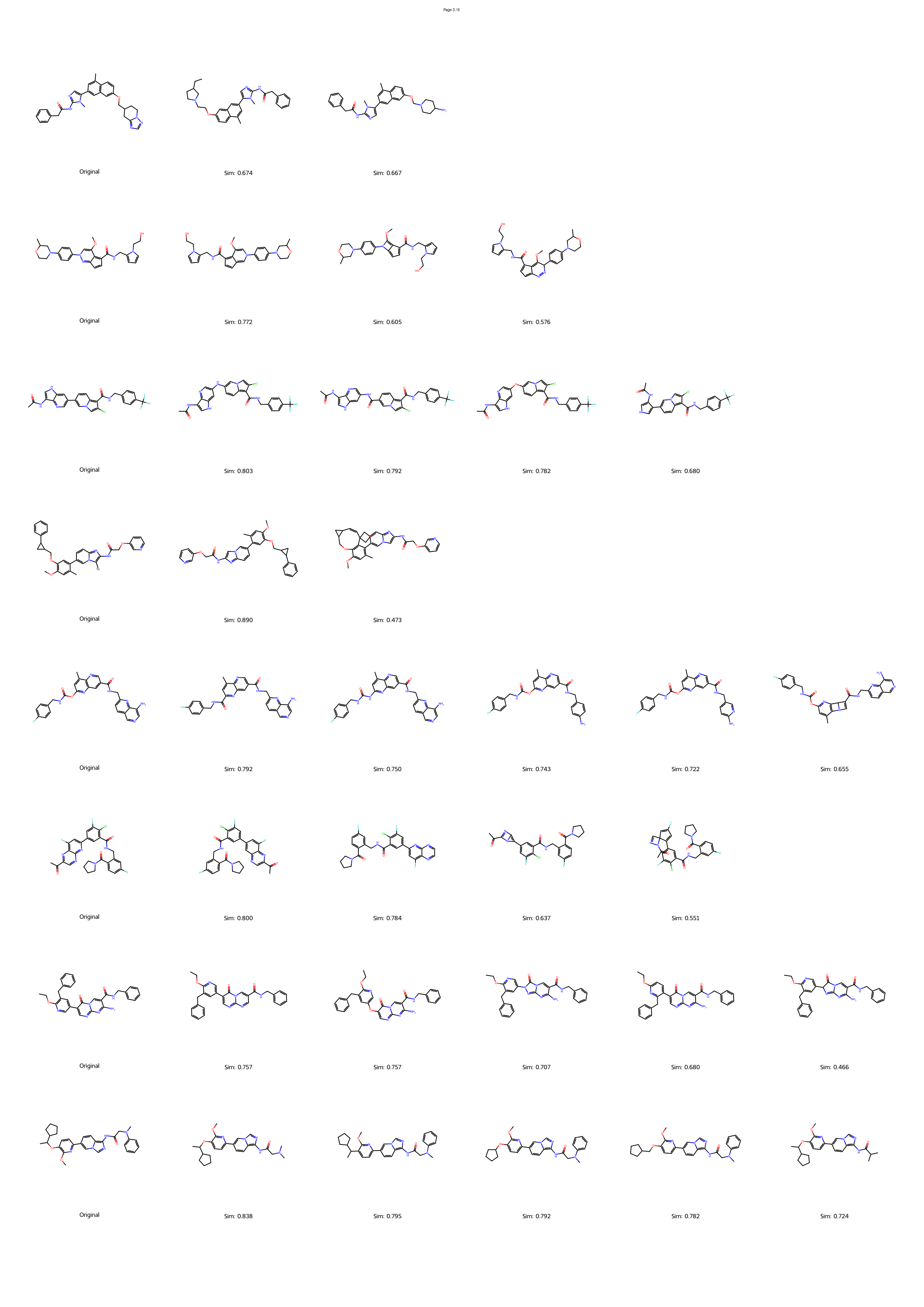
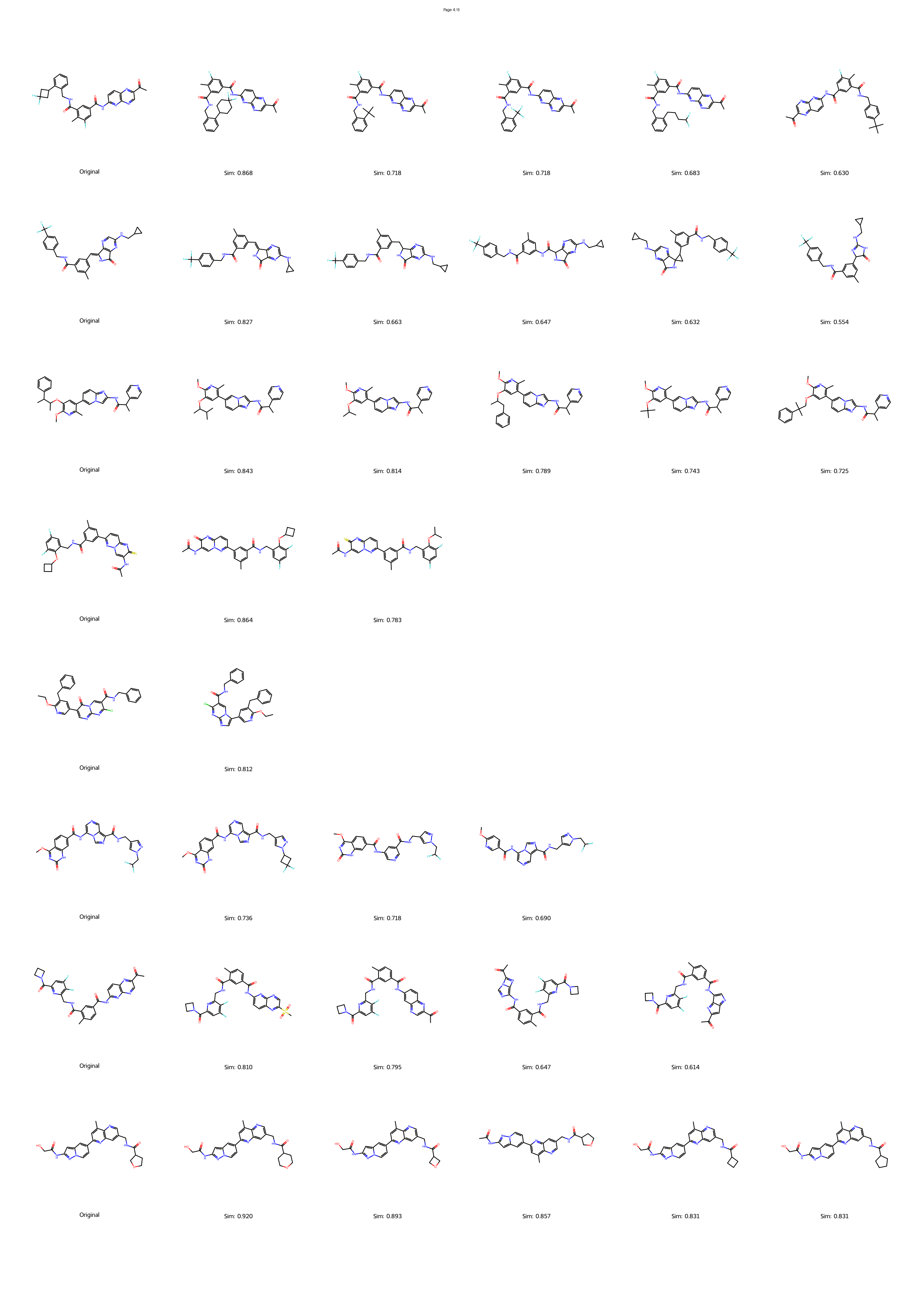
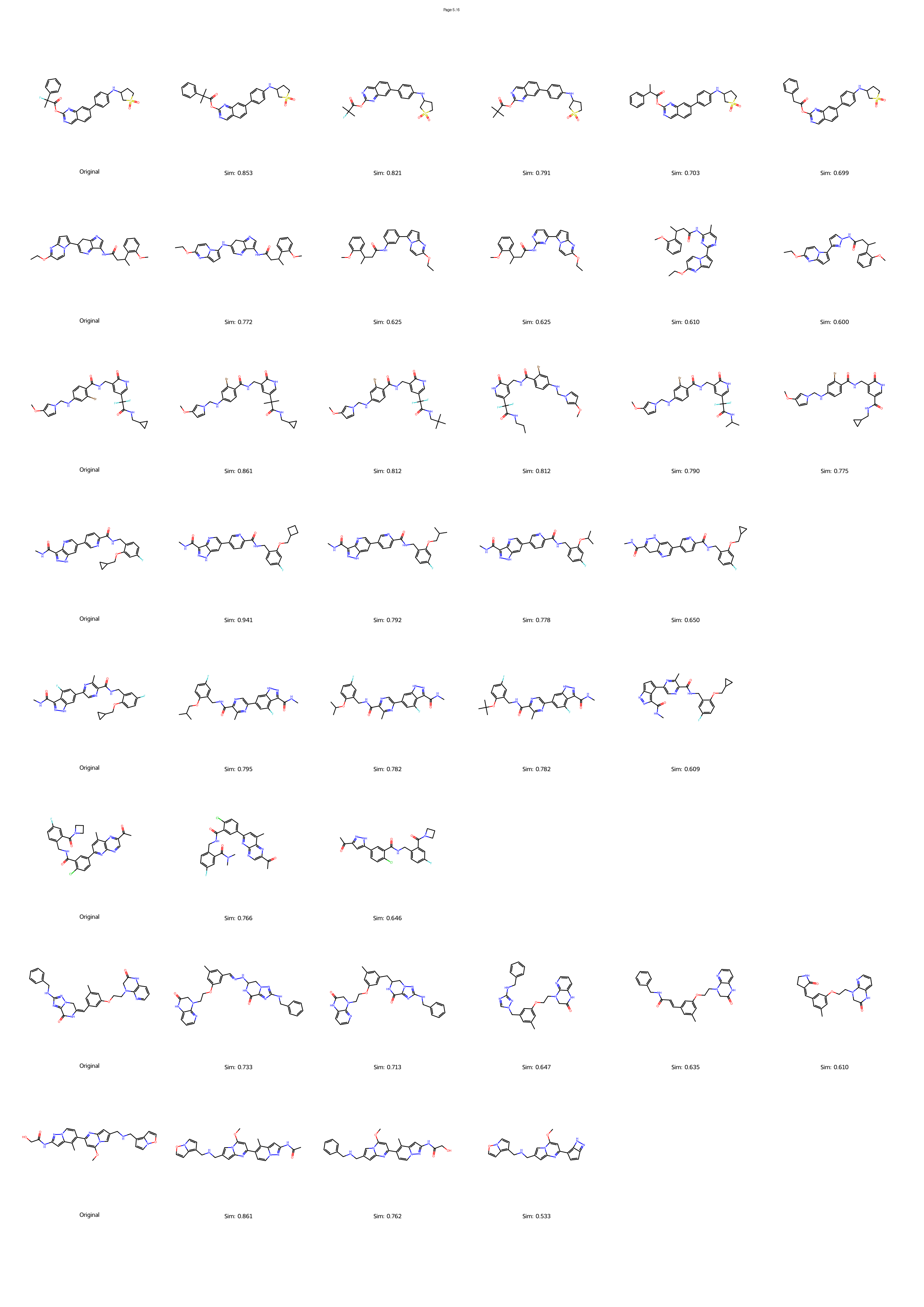
However, the distinction between a synthesizable and an unsynthesizable molecule is not always a vast chasm requiring projection into a distant chemical neighborhood. We observe that frequently, a minor and localized structural modification is sufficient to transform a challenging target into an accessible one. 25 This phenomenon is conceptually analogous to the well-established “activity cliffs” in medicinal chemistry, where a subtle change in a molecular structure can induce a dramatic shift in its biological activity. 26,27 This leads us to define a corresponding concept, the “synthesis cliff”, where a small, targeted structural edit can dramatically improve synthetic feasibility. Therefore, the central goal of this work is to develop a new approach that performs precise, minimal modifications to navigate this synthesis cliff, thereby salvaging promising designs without being confined to a limited repertoire of reaction templates.
The recent advent of Large Language Models (LLMs) offers a compelling new avenue to address this challenge. [28][29][30] Trained on vast corpora of scientific text, these models have developed an extensive, implicit knowledge base of chemical principles, reaction rules, and structural stability. Beyond rote knowledge, they exhibit emergent reasoning capabilities to formulate strategies and justify their decisions in human-readable language. 31 Despite this immense potential, a fundamental obstacle has hindered their direct application to molecular optimization. LLMs lack native proficiency in the strict, grammatical formalisms of chemical representations like SMILES. [32][33][34][35] This syntactic fragility means that when prompted to generate a complete molecule, they often produce invalid or chemically nonsensical outputs, leading to high failure rates and undermining their reliability for scientific tasks.
Herein, we introduce SynCraft, a novel reasoning-based framework designed to harness the chemical intelligence of LLMs while systematically circumventing their syntactic weaknesses. Instead of tasking the LLM with the direct generation of a full SMILES string, we reframe its role to that of a chemical strategist. Through a Chain-of-Thought (CoT) prompting strategy, 31 we guide the model to first reason about the synthetic liabilities within a molecule and then formulate a plan. This plan is not a new molecule, but rather a precise sequence of atom-level edits (e.g., DEL ATOM, ADD BOND). These instructions are then passed to a deterministic chemical toolkit for flawless execution, guaranteeing the validity of the final structure. 36 Through extensive benchmarking, we demonstrate that SynCraft significantly outperforms current state-of-the-art methods, particularly in generating synthesizable analogs with high structural similarity to the original input. Moving beyond quantitative simi
This content is AI-processed based on open access ArXiv data.