Authors: Nils Küchenmeister, Alex Ivliev, Markus Krötzsch
📝 Abstract
We present a new use of Answer Set Programming (ASP) to discover the molecular structure of chemical samples based on the relative abundance of elements and structural fragments, as measured in mass spectrometry. To constrain the exponential search space for this combinatorial problem, we develop canonical representations of molecular structures and an ASP implementation that uses these definitions. We evaluate the correctness of our implementation over a large set of known molecular structures, and we compare its quality and performance to other ASP symmetry-breaking methods and to a commercial tool from analytical chemistry. Under consideration in Theory and Practice of Logic Programming (TPLP).
💡 Deep Analysis
📄 Full Content
Mass spectrometry is a powerful technique to determine the chemical composition of a substance (De Hoffmann and Stroobant 2007). However, the mass spectrum of a substance does not reveal its exact molecular structure, but merely the possible ratios of elements in the compound and its fragments. To identify a sample, researcher may use commercial databases (for common compounds), or software tools that can discover molecular structures from the partial information available. The latter leads to a combinatorial search problem that is a natural fit for answer set programming (ASP). Molecules can be modeled as undirected graphs, representing the different elements and atomic bonds as node and edge labels, respectively. ASP is well-suited to encode chemical domain knowledge (e.g., possible number of bonds for carbon) and extra information about the sample (e.g., that it has an OH group), so that each answer set encodes a valid molecular graph.
Unfortunately, this does not work: a direct ASP encoding yields exponentially many answer sets for each molecular graph due to the large number of symmetries (automorphisms) in such graphs. For example, C 6 H 12 O admits 211 distinct molecule structures but leads to 111,870 answer sets. Removing redundant solutions and limiting the search to unique representations are common techniques used in the ASP community where they have motivated research on symmetry-breaking. Related approaches work by rewriting the ground program before solving, see (Drescher et al. 2011, sbass) and (Devriendt et al. 2016a;Devriendt and Bogaerts 2016, BreakID), or augmenting ASP programs with additional constraints learned from generated instances (Tarzariol et al. 2023, ilasp using sbass). Some methods also integrate symmetry-breaking with existing solvers (Devriendt et al. 2016b, idp3), or provide dedicated solvers (Khaled and Benhamou 2018, HC-asp). In addition to these general approaches, there are also methods that explicitly define symmetry-breaking constraints for undirected graphs (Codish et al. 2019). However, our experiments with some of these approaches still produced 10-10,000 times more answer sets than molecules even in simple cases.
We therefore develop a new approach that prevents symmetries in graph representations already during grounding, and use it as the core of an ASP-based prototype implementation for enumerating molecular structures based on partial chemical information. In Section 2, we explain the problem and our prototype tool from a user perspective. We then define the problem formally in Section 3, using an abstract notion of tree representations of molecular graphs that takes inspiration from the chemical notation SMILES (Weininger 1988). We then derive a new canonical representation for molecular graphs (Section 4) to guide our ASP implementation (Section 5). In Section 6, we evaluate the correctness, symmetry-breaking capabilities, and performance of our tool in comparison to other ASP-based approaches and a leading commercial software for analytical chemistry (Gugisch et al. 2015). We achieve perfect symmetry-breaking, i.e., the removal of all redundant solutions, for acyclic graph structures and up to three orders of magnitude reduction in answer sets for cyclic cases in comparison to other ASP approaches. Overall, ASP therefore appears to be a promising basis for this use case, and possibly for other use cases concerned with undirected graph structures.
Many mass spectrometers break up samples into smaller fragments and measure their relative abundance. The resulting mass spectrum forms a characteristic pattern, enabling inferences about the underlying sample. High-resolution spectra may contain information such as “the molecule has six carbon atoms” or “there is an OH group”, but cannot reveal the samples’s full molecular structure. In chemical analysis, we are looking for molecular structures that are consistent with the measured mass spectrum.
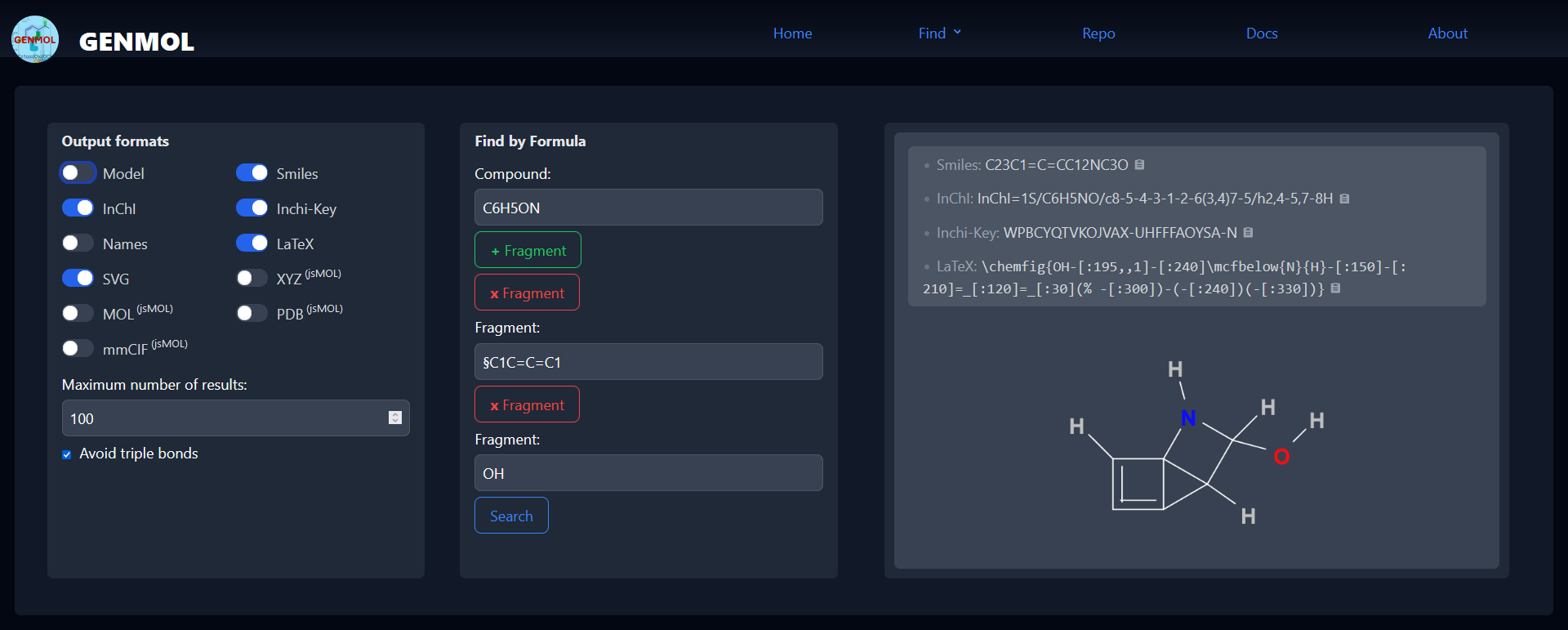
To address this task, we have developed Genmol, a prototype application for enumerating molecular structures for a given composition of fragments. It is available as a command-line tool and as a progressive web application (PWA), shown in Fig. 1. Genmol is implemented in Rust, with the web front-end using the Yew framework on top of a JSON-API, whereas the search for molecular structures is implemented in Answer Set Programming (ASP) and solved using clingo (Gebser et al. 2011). An online demo of Genmol is available for review at https://tools.iccl.inf.tu-dresden.de/genmol/
.
The screenshot shows the use of Genmol with a sum formula C 6 H 5 ON and two fragments as input. Specifying det