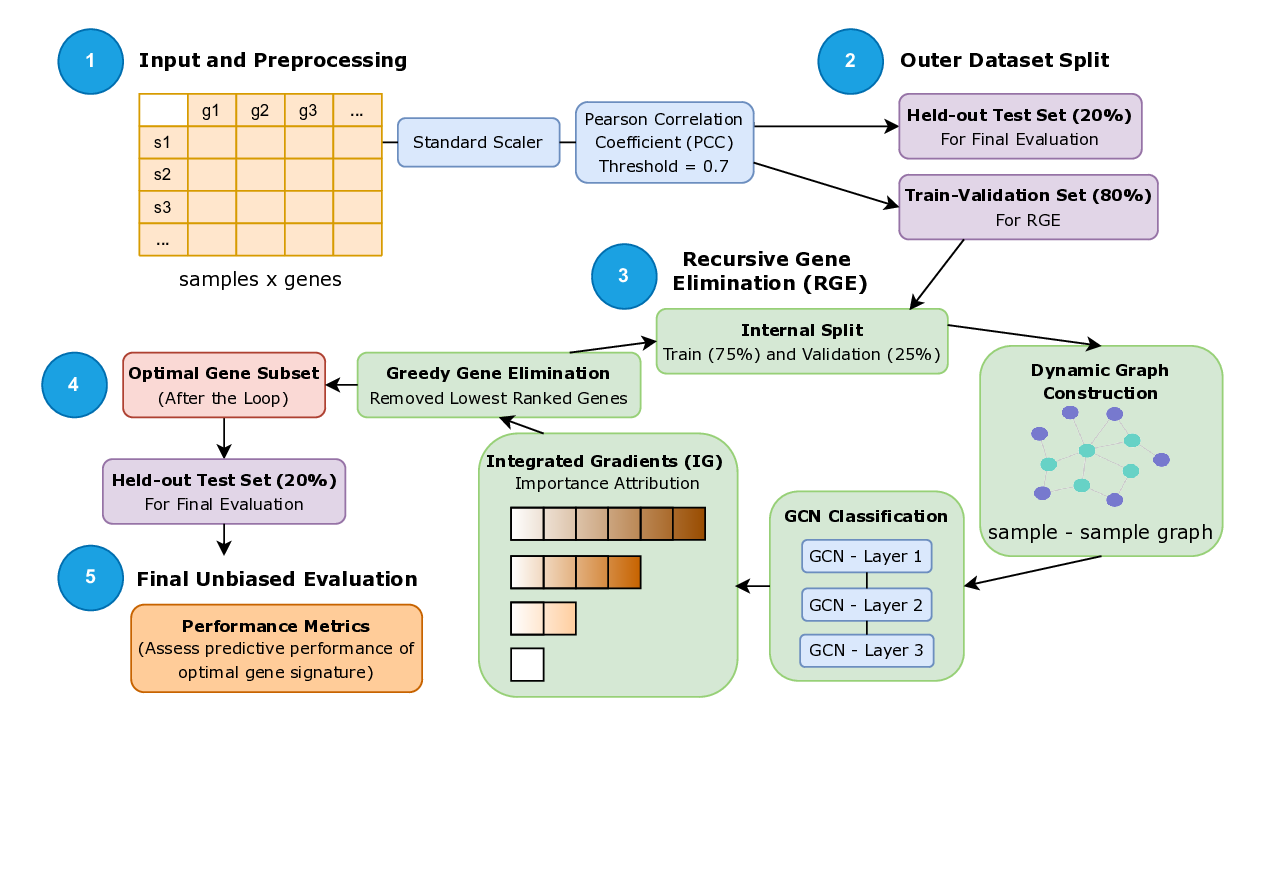
Early detection of cancer plays a key role in improving survival rates, but identifying reliable biomarkers from RNA-seq data is still a major challenge. The data are high-dimensional, and conventional statistical methods often fail to capture the complex relationships between genes. In this study, we introduce RGE-GCN (Recursive Gene Elimination with Graph Convolutional Networks), a framework that combines feature selection and classification in a single pipeline. Our approach builds a graph from gene expression profiles, uses a Graph Convolutional Network to classify cancer versus normal samples, and applies Integrated Gradients to highlight the most informative genes. By recursively removing less relevant genes, the model converges to a compact set of biomarkers that are both interpretable and predictive. We evaluated RGE-GCN on synthetic data as well as real-world RNA-seq cohorts of lung, kidney, and cervical cancers. Across all datasets, the method consistently achieved higher accuracy and F1-scores than standard tools such as DESeq2, edgeR, and limma-voom. Importantly, the selected genes aligned with well-known cancer pathways including PI3K-AKT, MAPK, SUMOylation, and immune regulation. These results suggest that RGE-GCN shows promise as a generalizable approach for RNA-seq based early cancer detection and biomarker discovery (https://rce-gcn.streamlit.app/ ).
💡 Deep Analysis
📄 Full Content
RGE-GCN: Recursive Gene Elimination with Graph Convolutional
Networks for RNA-seq based Early Cancer Detection
Shreyas Shendea, Varsha Narayanana, Vishal Fennb, Yiran Huangb, Dincer Goksulukc,
Gaurav Choudharyd,e,f, Melih Agrazd,e and Mengjia Xua,b
aDepartment of Computer Science, New Jersey Institute of Technology, Newark, 07102, NJ, USA
bDepartment of Data Science, New Jersey Institute of Technology, 07102, Newark, NJ, USA
cDepartment of Biostatistics, Sakarya, 54187, Turkiye
dDivision of Cardiology, Brown University Health, Providence, 02903, RI, USA
eDepartment of Medicine, Warren Alpert Medical School of Brown University, Providence, RI, 02903, USA
fVA Providence Healthcare System, Providence, RI, 02903, USA
A R T I C L E I N F O
Keywords:
Graph Neural Networks (GNN)
Differentially Expressed Genes (DEGs)
RNA-Sequence
Integrated Gradients (IG)
A B S T R A C T
Early detection of cancer plays a key role in improving survival rates, but identifying reliable
biomarkers from RNA-seq data is still a major challenge. The data are high-dimensional, and
conventional statistical methods often fail to capture the complex relationships between genes. In this
study, we introduce RGE-GCN (Recursive Gene Elimination with Graph Convolutional Networks),
a framework that combines feature selection and classification in a single pipeline. Our approach
builds a graph from gene expression profiles, uses a Graph Convolutional Network to classify cancer
versus normal samples, and applies Integrated Gradients to highlight the most informative genes.
By recursively removing less relevant genes, the model converges to a compact set of biomarkers
that are both interpretable and predictive. We evaluated RGE-GCN on synthetic data as well as
real-world RNA-seq cohorts of lung, kidney, and cervical cancers. Across all datasets, the method
consistently achieved higher accuracy and F1-scores than standard tools such as DESeq2, edgeR, and
limma-voom. Importantly, the selected genes aligned with well-known cancer pathways including
PI3K–AKT, MAPK, SUMOylation, and immune regulation. These results suggest that RGE-GCN
shows promise as a generalizable approach for RNA-seq based early cancer detection and biomarker
discovery (https://rce-gcn.streamlit.app/).
1. Introduction
Genomic data analytics has become increasingly critical
in advancing our understanding of cancer, particularly in
detecting the disease at an early stage [9]. RNA sequenc-
ing (RNA-seq) enables high-resolution examination of gene
expression profiles across diverse samples, making it a pow-
erful tool for biomarker discovery. However, the inherently
high dimensionality of RNA-seq data involving simultane-
ous measurements of tens of thousands of genes presents sig-
nificant computational and statistical challenges. Accurately
identifying differentially expressed genes (DEGs), which
show meaningful differences between healthy and cancer-
ous samples, is therefore of critical importance. Effective
DEG selection not only reduces complexity by highlight-
ing biologically relevant genes but also enhances model
interpretability and strengthens classification performance.
Ultimately, accurate DEG identification directly contributes
to the discovery of reliable biomarkers for early cancer de-
tection, thereby improving patient outcomes and advancing
early diagnostic tools.
High-dimensional gene expression data, characterized
by a large number of features and a limited number of sam-
ples, increase the risk of overfitting and exacerbate computa-
tional complexity in classical machine learning approaches
∗Corresponding author
mx6@njit.edu (M. Xu)
ORCID(s):
[28]. To address these limitations, Graph Neural Networks
(GNNs) have emerged as powerful tools, offering a struc-
tured framework to capture both co-expression patterns
and biological interactions. Recent efforts can be broadly
grouped into two categories: (1) graph-based models and (2)
specialized architectures designed for targeted biomedical
applications. (1) Graph-based models. Within the graph
domain, comparative studies such as Alharbi et al. [2]
evaluated GCN, GAT, and GTN architectures for multi-
omics cancer classification, demonstrating the benefit of
regularized feature reduction. Similarly, Li and Nabavi [18]
proposed a heterogeneous GNN to integrate inter and intra-
omics relationships. However, these approaches are largely
centered on multi-omics integration and often depend on
preselected feature sets, which constrains their applicability
when learning directly from single-omics RNA-seq data. In
addition, Wang et al. [32] introduced scGNN, a graph-based
framework for single-cell transcriptomics that effectively
models gene–gene and cell–cell dependencies, while Mao
et al. [3] framed gene regulatory networks (GRN) recon-
struction as a link-prediction task (GNNLink), using a GCN-
based encoder over prior TF–gene graphs and scRNA-seq
features to recover regulatory edges; across seven scRNA-
seq datasets and multiple