Reliable evaluation of protein structure predictions remains challenging, as metrics like pLDDT capture energetic stability but often miss subtle errors such as atomic clashes or conformational traps reflecting topological frustration within the protein folding energy landscape. We present CODE (Chain of Diffusion Embeddings), a self evaluating metric empirically found to quantify topological frustration directly from the latent diffusion embeddings of the AlphaFold3 series of structure predictors in a fully unsupervised manner. Integrating this with pLDDT, we propose CONFIDE, a unified evaluation framework that combines energetic and topological perspectives to improve the reliability of AlphaFold3 and related models. CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT's 0.33 (a relative improvement of 148\%). CONFIDE significantly enhances the reliability of quality evaluation in molecular glue structure prediction benchmarks, achieving a Spearman correlation of 0.73 with RMSD, compared to pLDDT's correlation of 0.42, a relative improvement of 73.8\%. Beyond quality assessment, our approach applies to diverse drug design tasks, including all-atom binder design, enzymatic active site mapping, mutation induced binding affinity prediction, nucleic acid aptamer screening, and flexible protein modeling. By combining data driven embeddings with theoretical insight, CODE and CONFIDE outperform existing metrics across a wide range of biomolecular systems, offering robust and versatile tools to refine structure predictions, advance structural biology, and accelerate drug discovery.
The advent of deep learning in structural biology, epitomized by AlphaFold3 (AF3), has transformed our ability to predict the three-dimensional structures of biomolecules including proteins, nucleic acids, and their complexes [1]. AF3 often achieves nearexperimental accuracy across a broad range of targets, accelerating progress in structural biology and drug development. Yet, despite these advances, the internal mechanisms by which AF3 generates its predictions, and the reliability of its self-assessed confidence, remain incompletely understood.
AF3 reports a predicted Local Distance Difference Test (pLDDT) score to quantify structural confidence. While pLDDT generally correlates with model accuracy, it fails in critical cases involving complex molecular assemblies or atypical conformations. Similar to other large-scale generative models, AF3 can produce hallucination, i.e. structures with deceptively high confidence but substantial deviations from experimentally determined conformations. This high-confidence/low-accuracy paradox undermines downstream applications, particularly in drug discovery, where misleading structural models may misdirect experimental resources. A deeper understanding of AF3’s generative process is therefore crucial for mitigating hallucinations and improving reliability, especially in data-scarce scenarios.
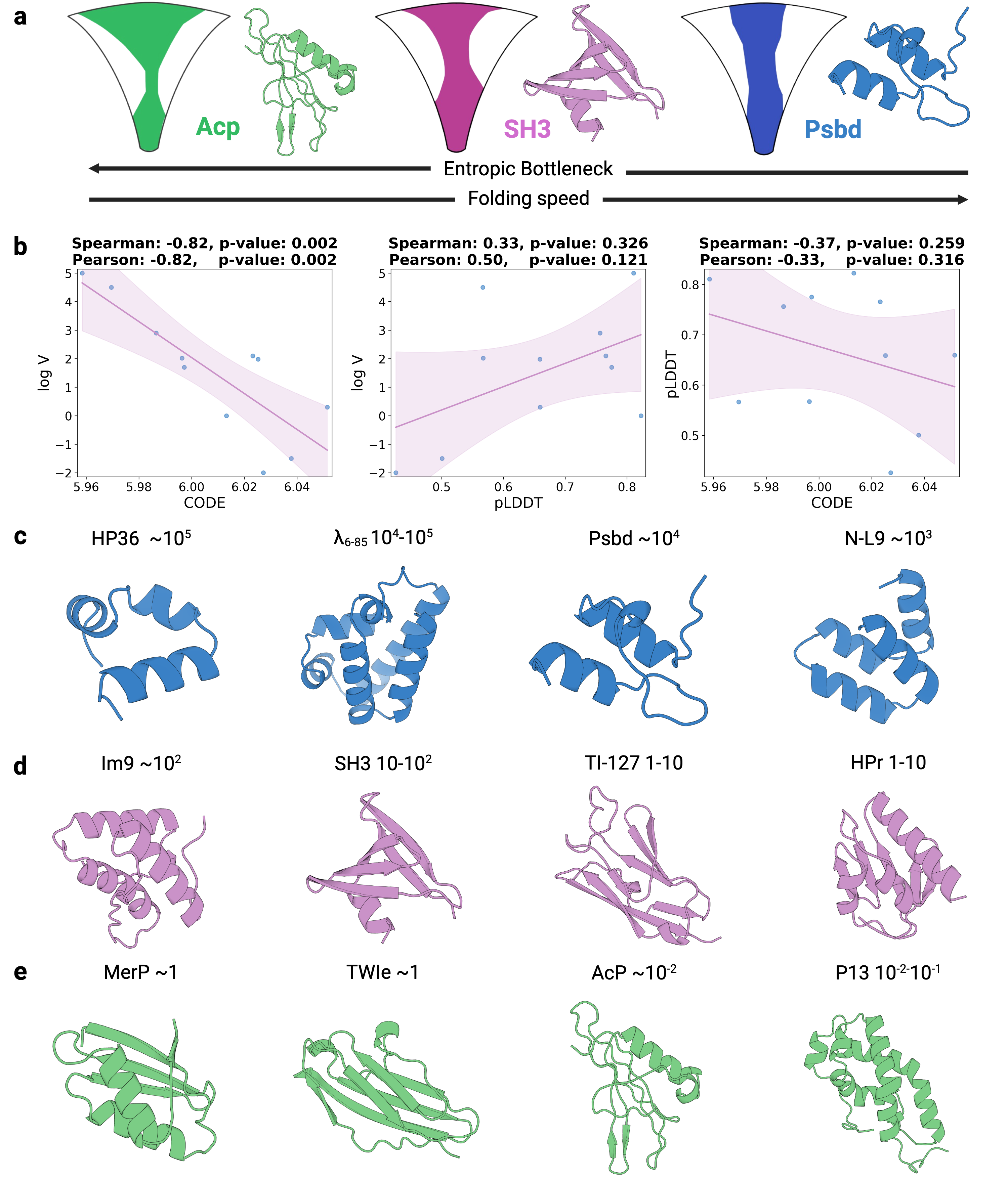
Classical interpretations of AlphaFold draw on protein-folding energy landscape theory. AlphaFold2 (AF2) was hypothesized to learn an energy function, using coevolutionary information from multiple sequence alignments (MSAs) to identify approximate global minima, followed by refinement through its structure module [2]. While this framework helps explain how AF2 resolves energetic frustration, it does not account for the additional constraints imposed by protein topology. Protein folding also reflects topological frustration, which arises from chain connectivity, conformational entropy losses, and the need to traverse rugged folding funnels without becoming trapped in local minima [3][4][5]. These topological constraints profoundly shape folding pathways and increase structural complexity, yet these intricate details of folding process are not well captured by existing data-driven methods.
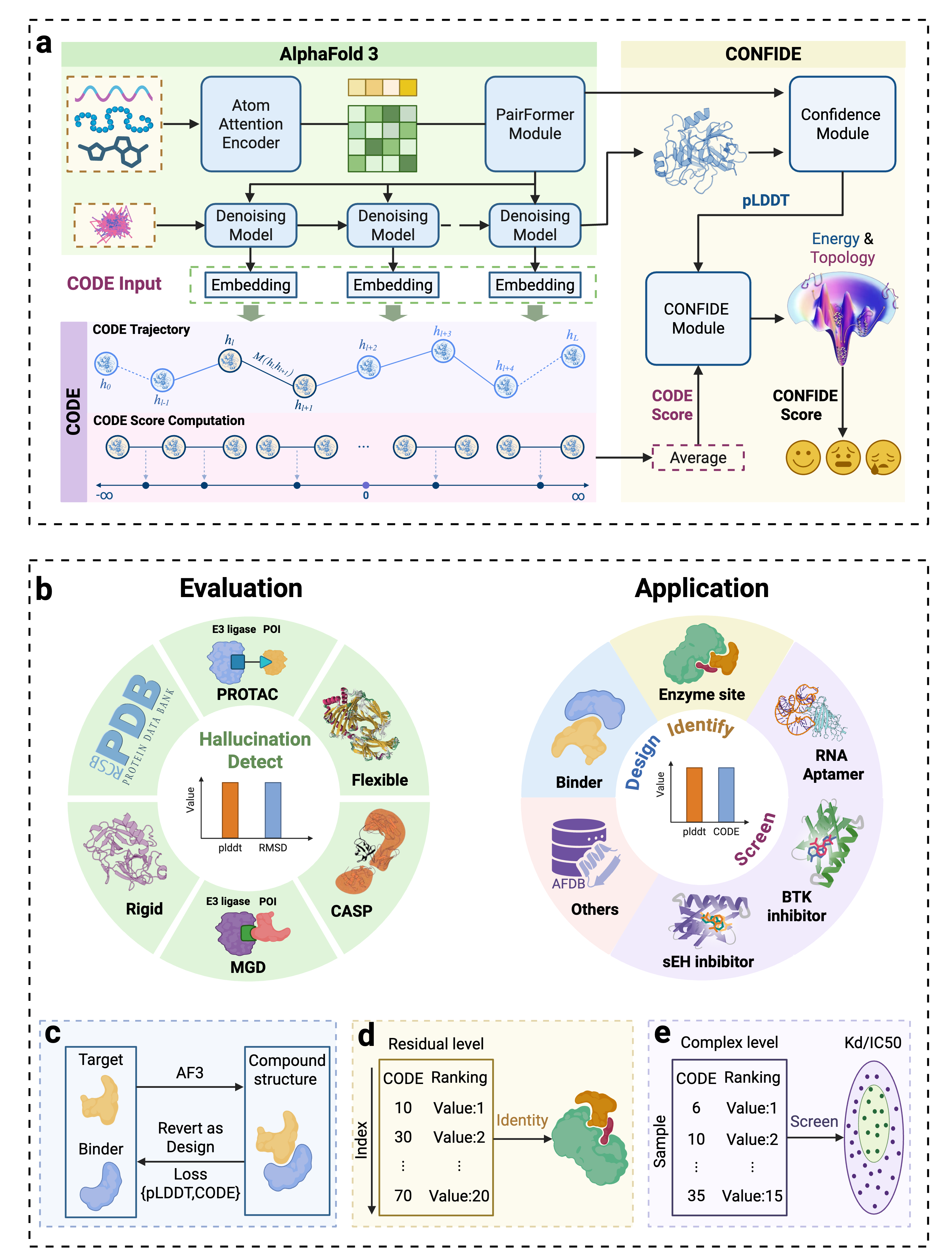
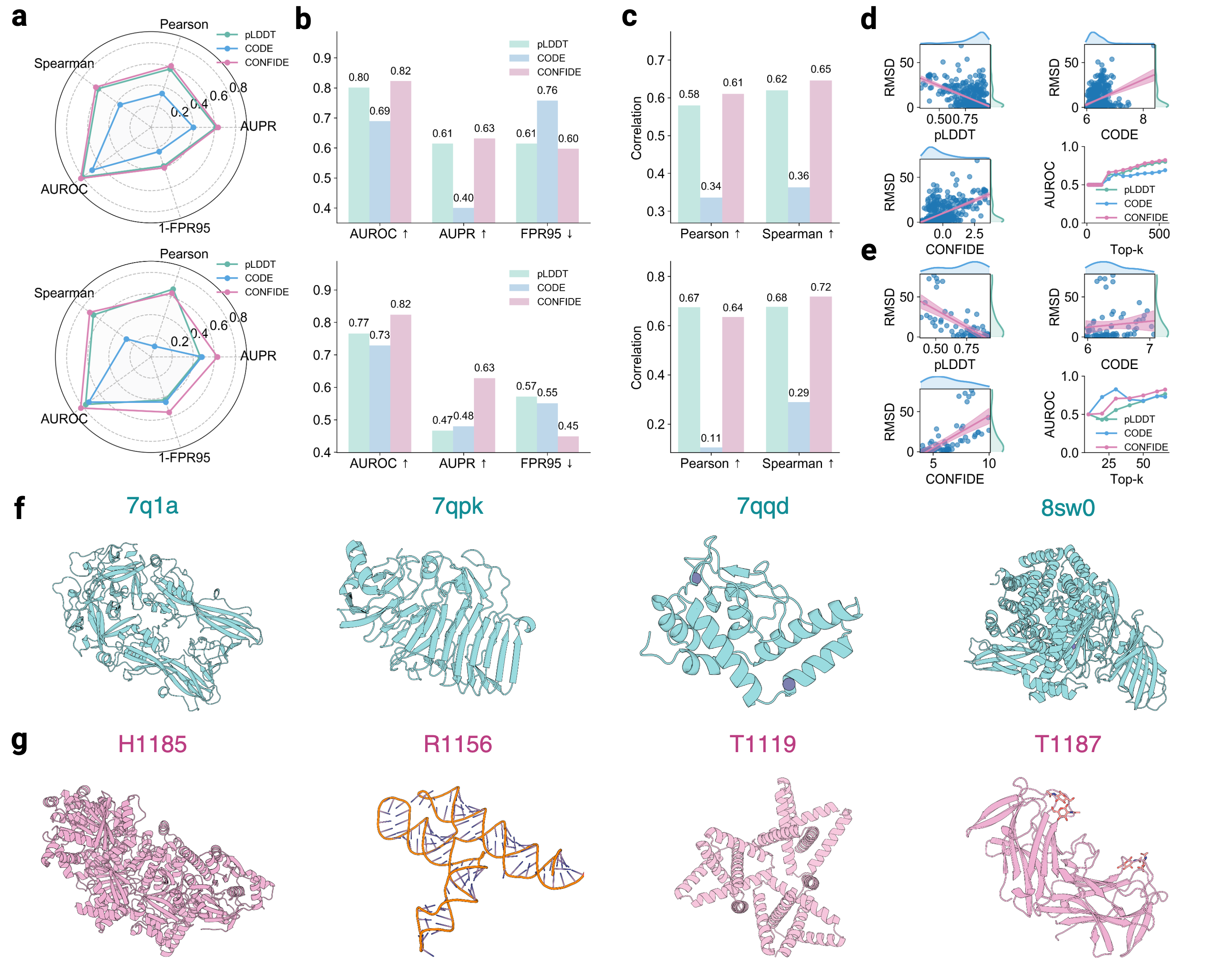
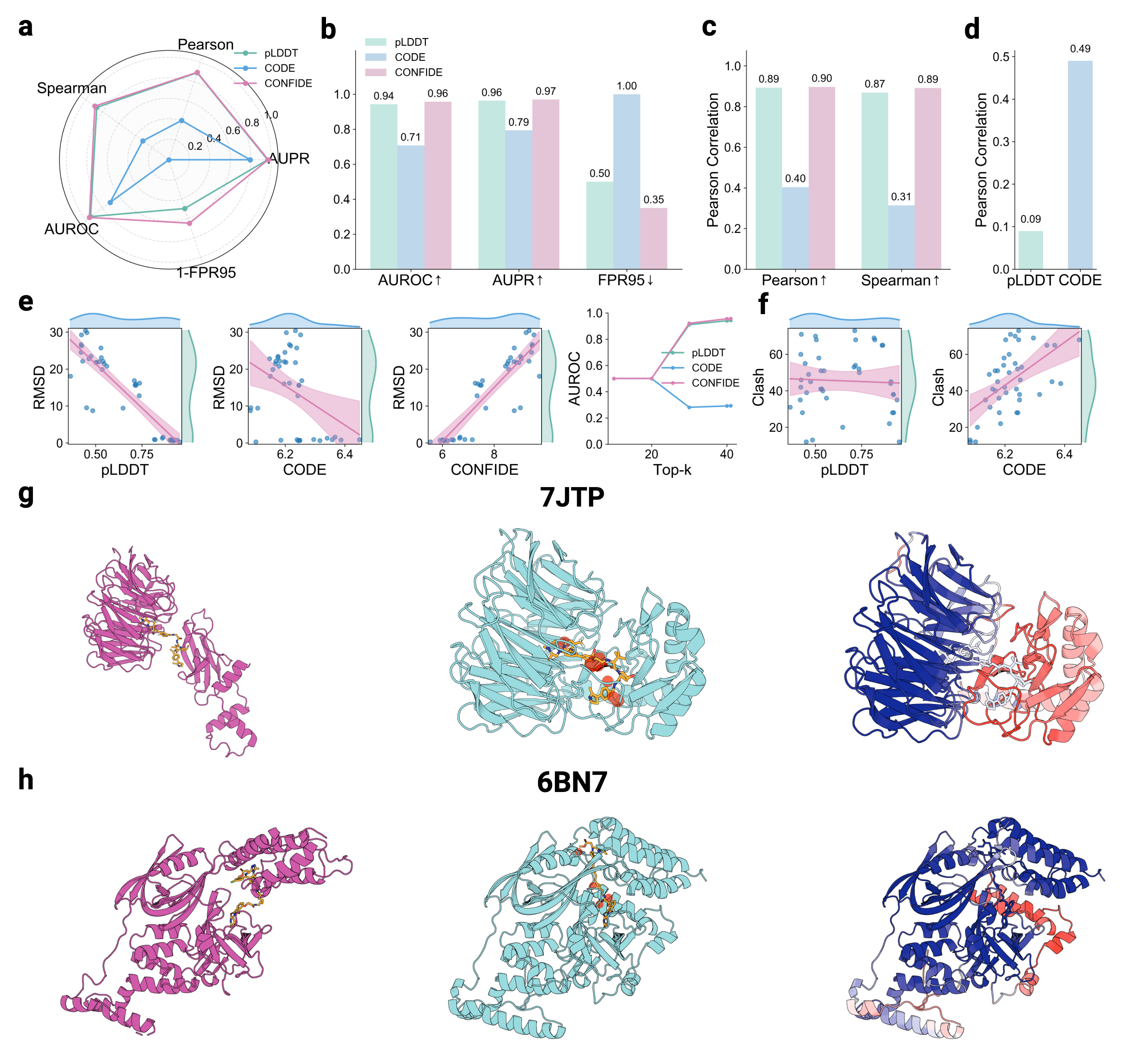
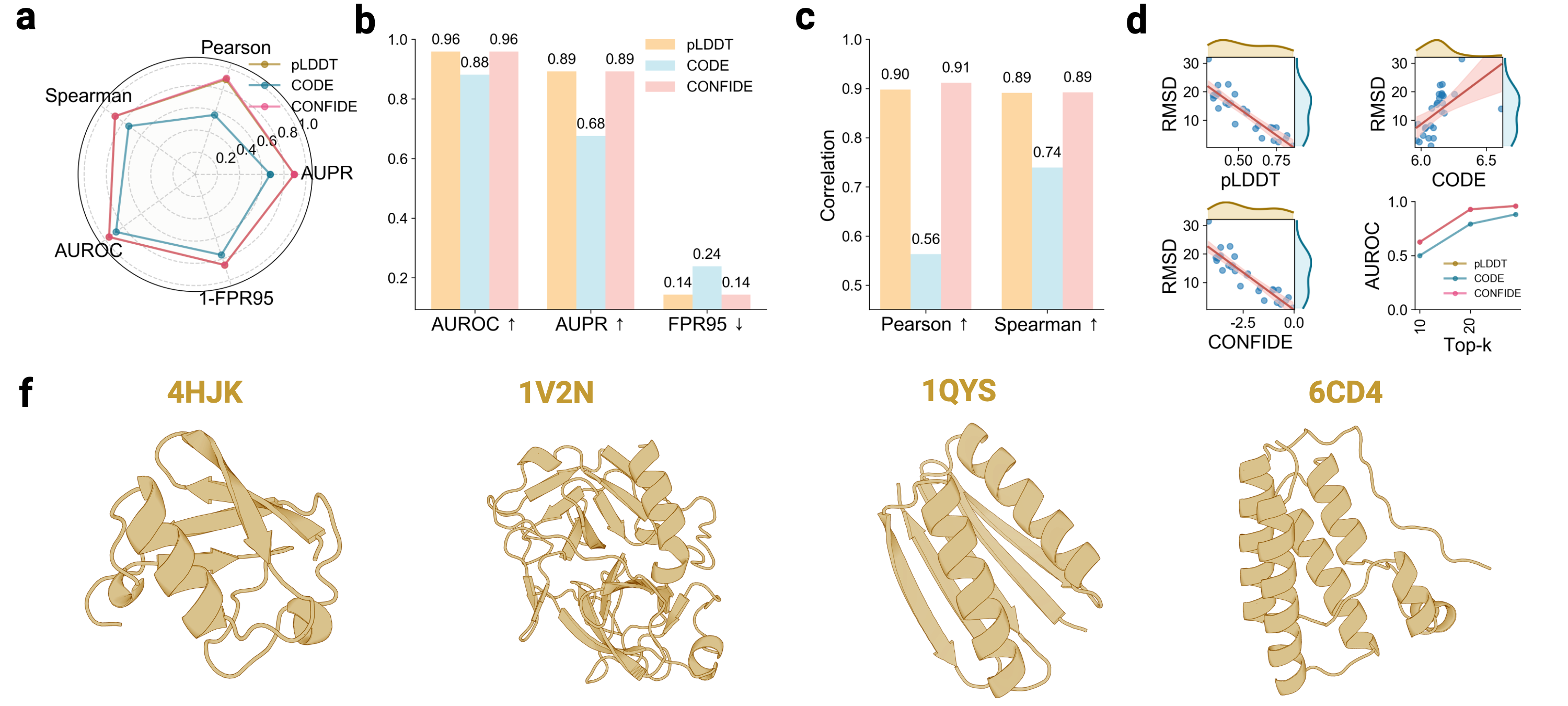
Building upon these insights, we investigate whether the generative process of the AlphaFold3 series of structure predictors can be understood more comprehensively through the lens of energy landscape theory. Here, we introduce a theoretical framework for interrogating the predictions of the AlphaFold3 series of structure predictors by quantifying topological frustration. Inspired by chain-of-thought reasoning in large language models (LLMs), we formalize the progressive latent representations within the diffusion-based structure module of the AlphaFold3 series of structure predictors as the Chain of Diffusion Embeddings (CODE) [6,7]. We hypothesize that these progressively refined embeddings capture long-range inter-residue relationships reflective of backbone constraints and topological complexity [8]. As mentioned above, these subtle structural constraints cannot be easily recognized by more localized metrics such as pLDDT. To validate this, we conducted proof-of-concept experiments demonstrating that CODE strongly correlates with protein folding rates driven by topological frustration, achieving a correlation of 0.82 compared to pLDDT’s 0.33 (a relative improvement of 148%).
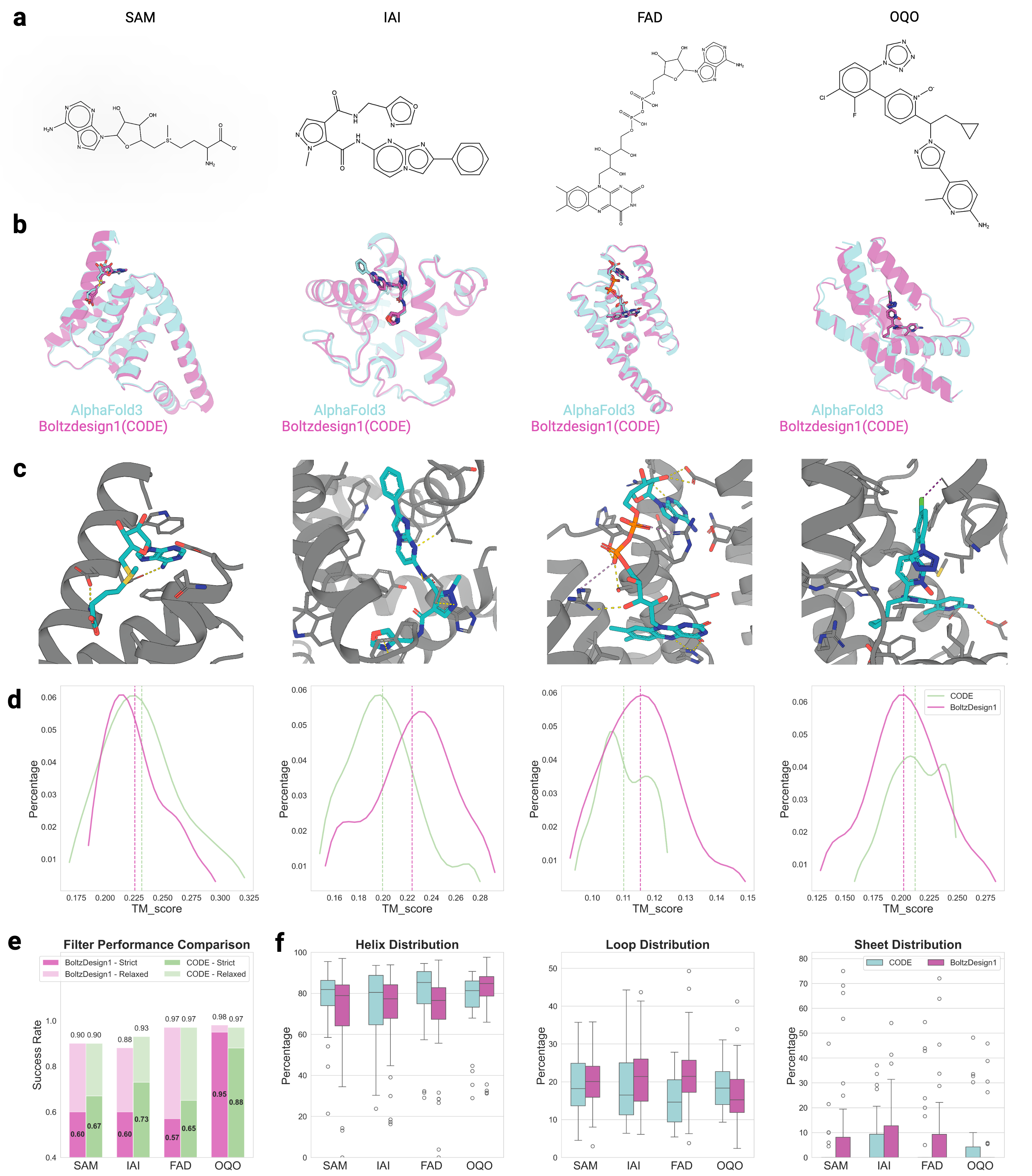
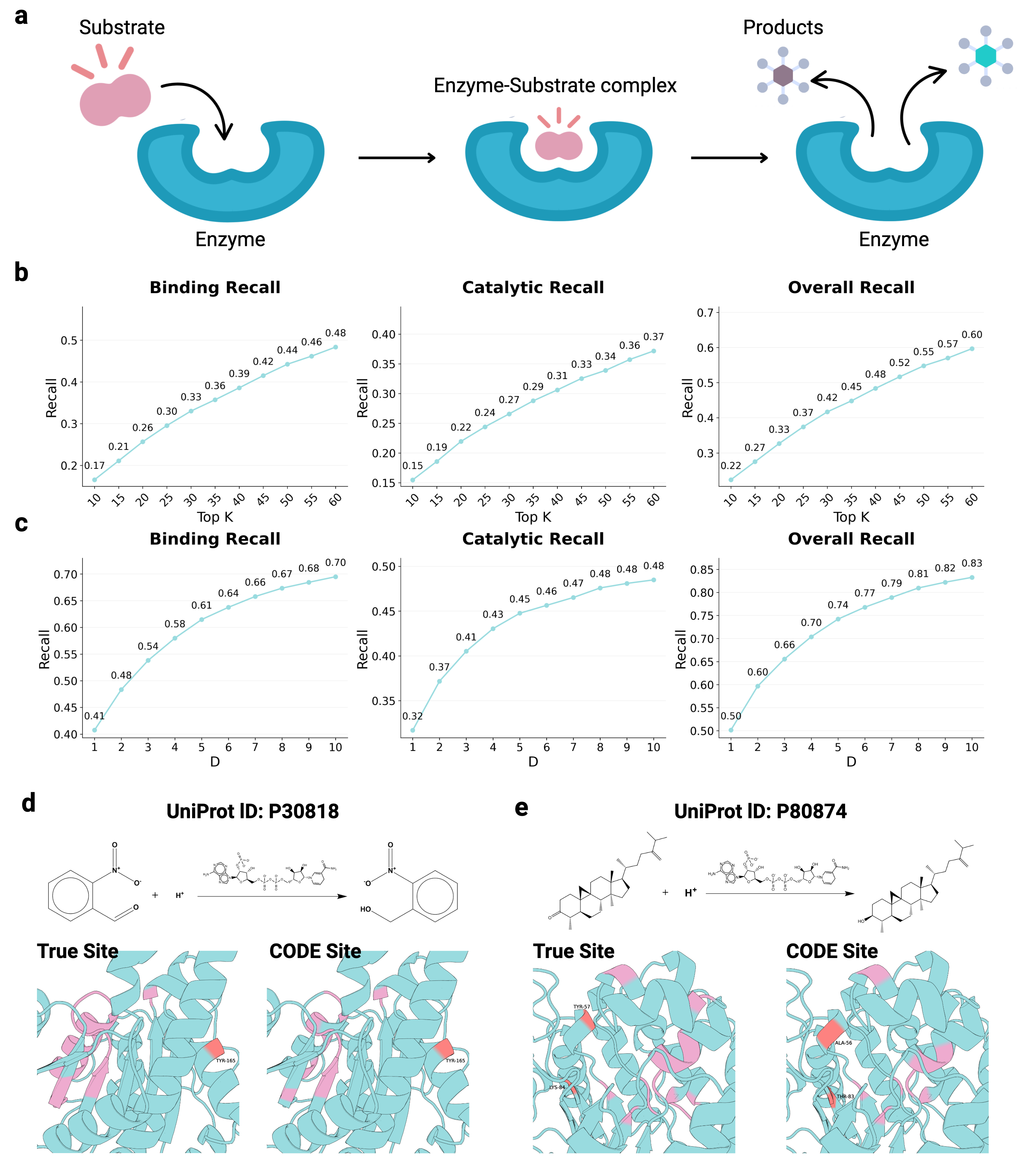
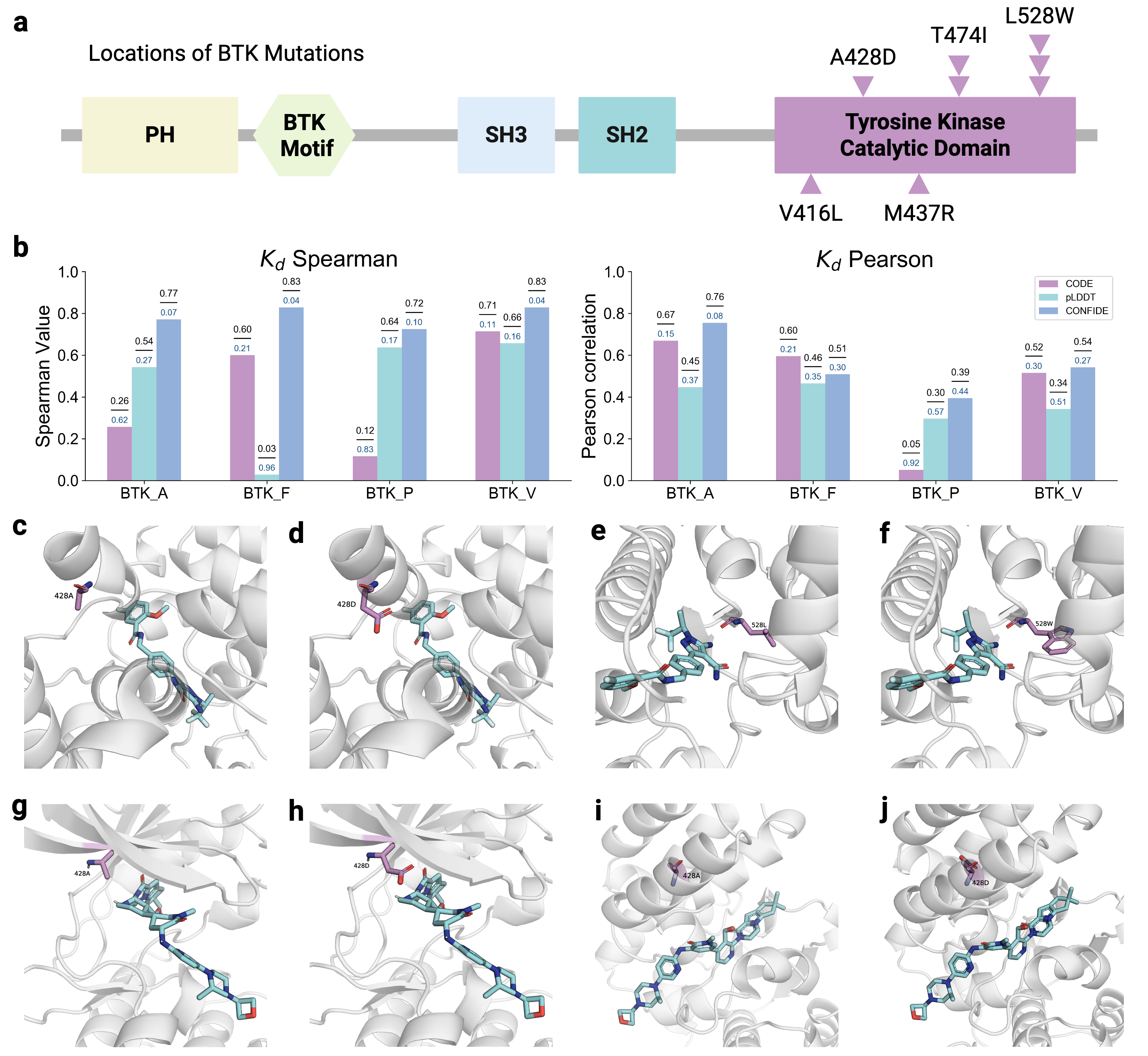
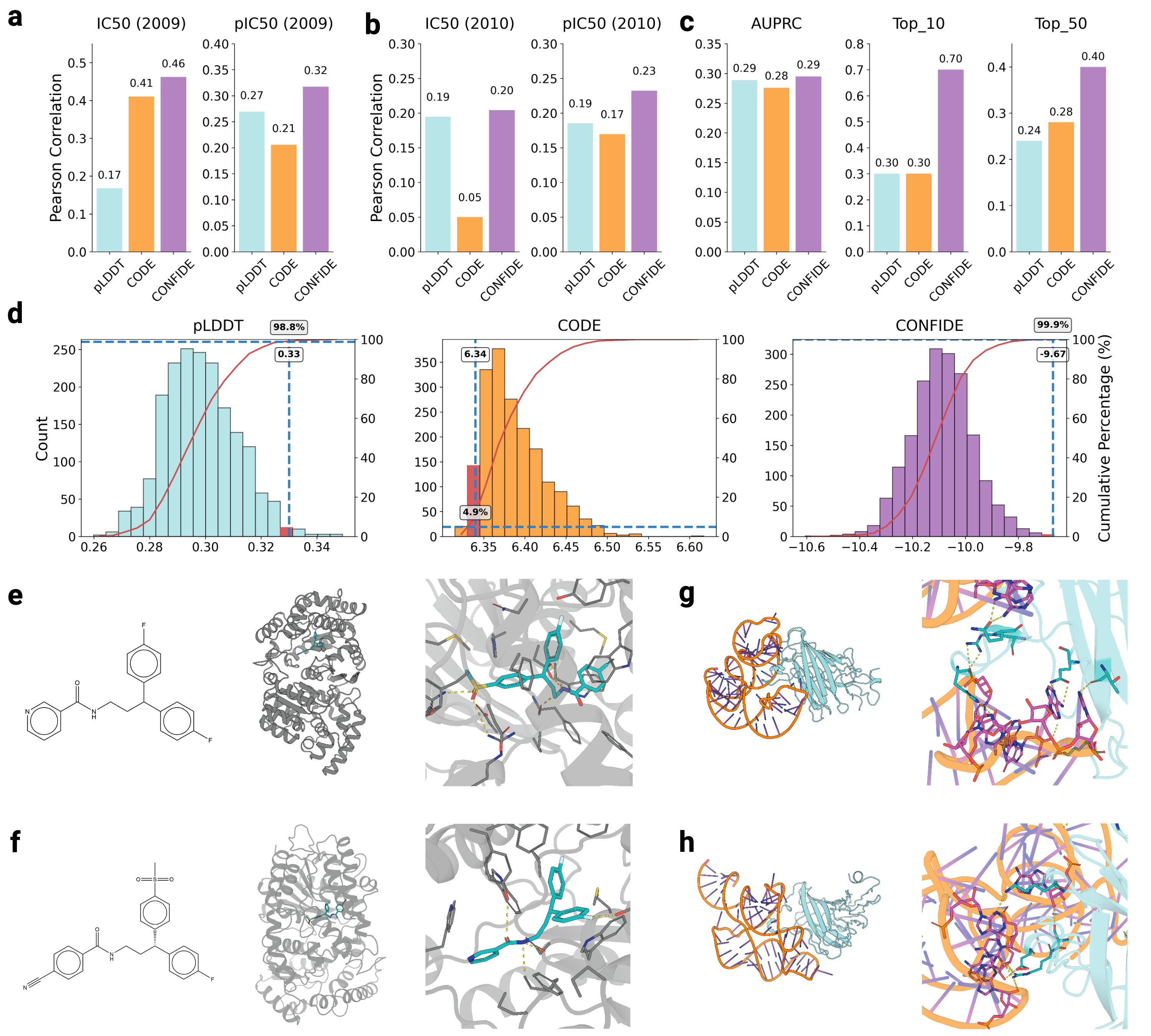
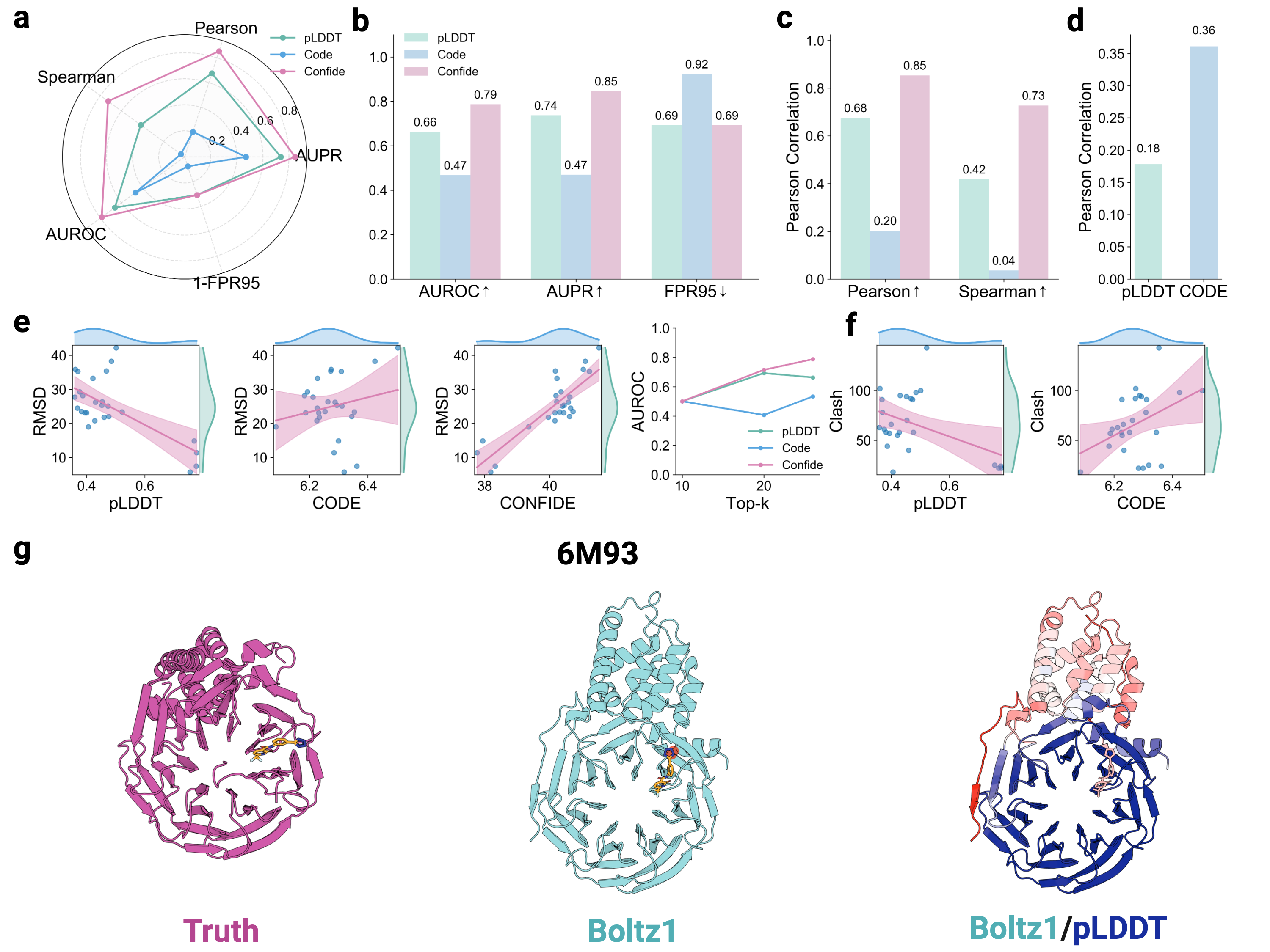
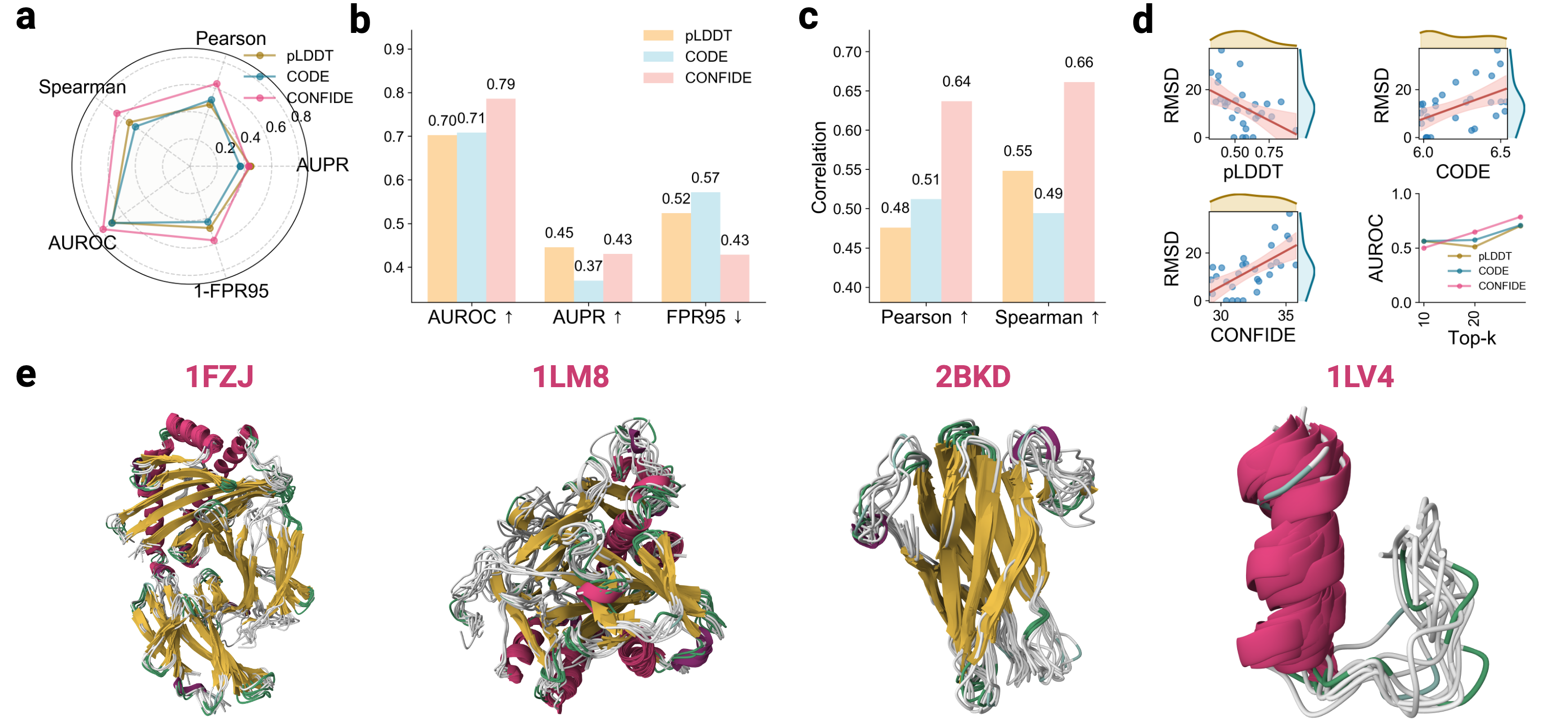
Recognizing the complementarity between CODE and pLDDT, we develop CONFIDE (CONFIdence-integrated Diffusion Embedding), a self-evaluating metric that unifies energetic (pLDDT) and topological (CODE) perspectives to provide a more robust assessment of structural predictions. Across diverse benchmarks-including ternary complexes, flexible and rigid proteins, and other challenging datasets-CONFIDE achieves superior performance compared to pLDDT, with particularly notable improvements in molecular-glue complexes (31% Spearman correlation enhancement). Furthermore, in downstream applications, CONFIDE operates in a fully unsupervised manner to guide binder design, enzyme catalytic site identification, mutation-induced affinity prediction, and inhibitor/nucleic acid aptamer screening, achieving excellent results across all tasks. Notably, CONFIDE improves the success rate of binder design for IAI by 13%. Additionally, CONFIDE accurately detects affinity changes from resistance mutations in the BTK protein against Fenebrutinib, with a Spearman correlation of 0.83, far exceeding pLDDT’s negligible 0.03.
By integrating embedding-based reasoning with theoretical insights into protein folding, CODE and CONFIDE provide a unified, interpretable framework for detecting structural hallucinations and improving model reliability. Addressing a key limitation in diffusion-based 3D generative modeling-widely used in structural biology, materials science, and engineering [9]-this framework universally applies to computational models with
This content is AI-processed based on open access ArXiv data.