강한 상관계 시스템을 위한 전이 가능한 딥 QMC 기반 ab initio 구조 최적화 프레임워크
본 논문은 전이 가능한 딥러닝 변분 몬테카를로(VMC) 파동함수를 활용해, 강한 전자 상관을 보이는 시스템의 잠재에너지면(PES)을 고정밀도로 탐색하는 새로운 방법을 제시한다. 핵 좌표를 지속적으로 샘플링하면서 파동함수를 학습하고, 에너지·힘·헤시안을 가우시안 프로세스 회귀(GPR)로 보정해 비용 효율적인 구조 최적화, 전이 상태 탐색, 최소 에너지 경로(MEP) 계산을 가능하게 한다.
저자: P. Bernát Szabó, Zeno Schätzle, Frank Noé
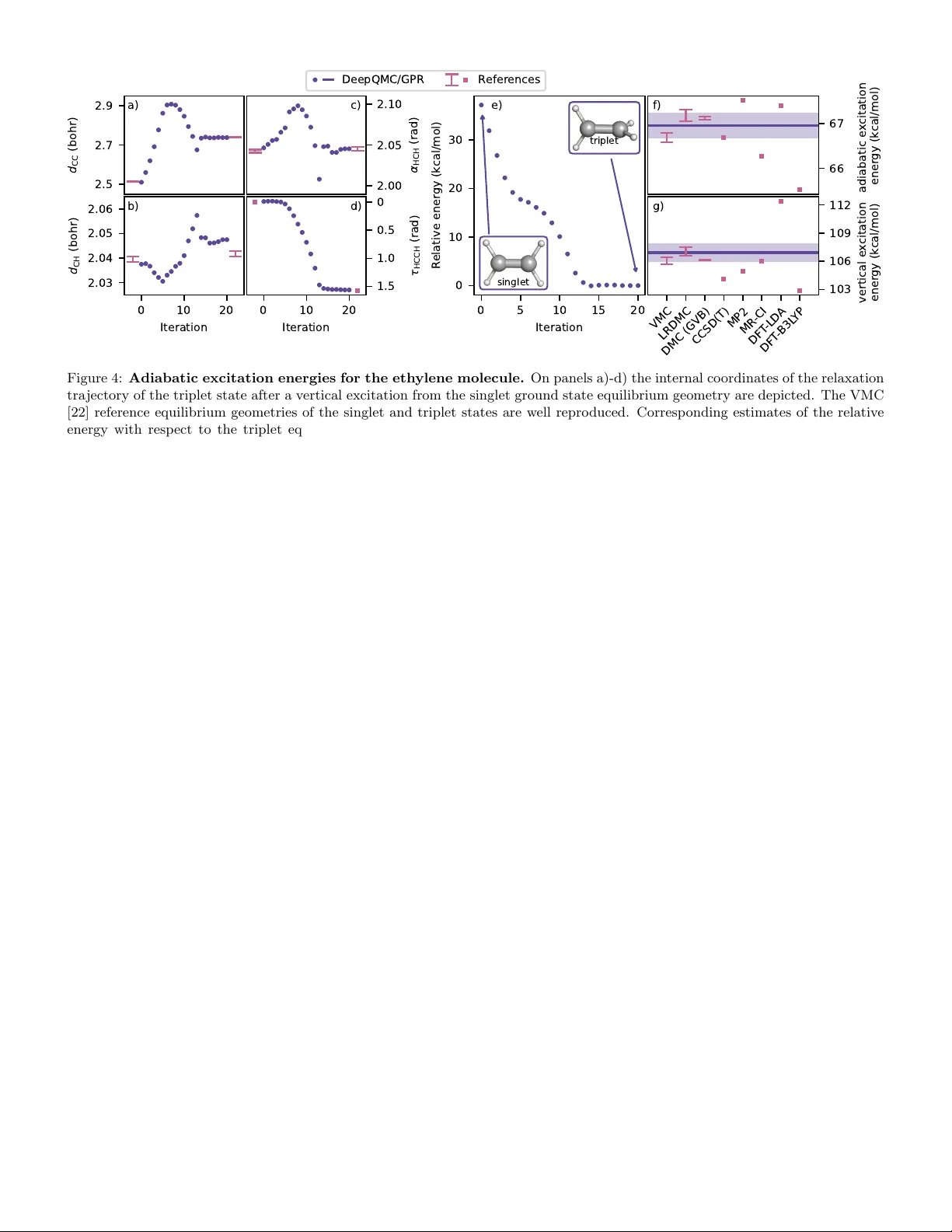
본 논문은 강하게 상관된 전자 시스템의 잠재에너지면(PES)을 고정밀도로 탐색하기 위한 새로운 ab initio 구조 최적화 프레임워크를 제시한다. 기존 양자화학 방법은 일반적으로 하나의 핵 좌표에 대해 전자 구조를 해결하고, 그 결과를 바탕으로 별도의 샘플링 과정을 거쳐 PES를 구축한다. 그러나 이러한 접근은 강한 전자 상관이나 다중 기준(ground / excited) 상태를 포함하는 경우, 계산 비용이 급격히 상승하고 정확도가 떨어지는 문제가 있다.
저자들은 이러한 한계를 극복하기 위해 전이 가능한 딥러닝 변분 몬테카를로(VMC) 파동함수 Ψθ(r|R)를 도입한다. 여기서 r은 전자 좌표, R은 핵 좌표이며, 파라미터 θ는 신경망 가중치이다. 파동함수는 핵 좌표 분포 ρ(R) 위에서 동시에 최적화되며, ρ(R)는 결합 길이, 각도, 비틀림 등을 포함하는 화학적으로 의미 있는 영역을 균일하게 샘플링한다. 학습 과정에서는 전자 배치를 무작위로 선택하고, Markov chain Monte Carlo와 space‑warp 기법을 결합해 전자 좌표를 핵 변형에 맞게 효율적으로 업데이트한다. 이로써 단일 파동함수 학습 비용이 개별 단일‑포인트 VMC와 비슷한 수준이면서, 광범위한 구조에 대해 일관된 에너지와 힘을 제공한다.
학습된 전이 가능한 파동함수는 직접적인 에너지·힘 추정에 사용될 수 있다. 하지만 Monte Carlo 추정은 본질적으로 노이즈가 존재하므로, 저자들은 두 가지 보완책을 적용한다. 첫째, 최신 zero‑variance 힘 추정기를 사용해 통계적 편향을 최소화한다. 둘째, 샘플링된 에너지와 힘 데이터를 기반으로 가우시안 프로세스 회귀(GPR)를 수행한다. GPR은 관측값 Y =
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기