희소성을 활용한 다중 유전자 변이 탐색 알고리즘
** 본 논문은 암 조직에서 관찰되는 높은 희소성을 이용해 다중 유전자 변이 조합을 효율적으로 탐색하는 Pruned Depth‑First Search(P‑DFS) 알고리즘을 제안한다. P‑DFS는 후보 조합을 깊이‑우선으로 탐색하면서 불가능한 서브트리를 조기에 제거하고, 이후 가중 집합 커버(Weighted Set Cover) 단계에서 가장 차별적인 조합을 선택한다. 대규모 슈퍼컴퓨터 실험에서 4‑hit 조합에 대해 90‑98%의 탐색 감소…
저자: Ritvik Prabhu, Emil Vatai, Bernard Moussad
**
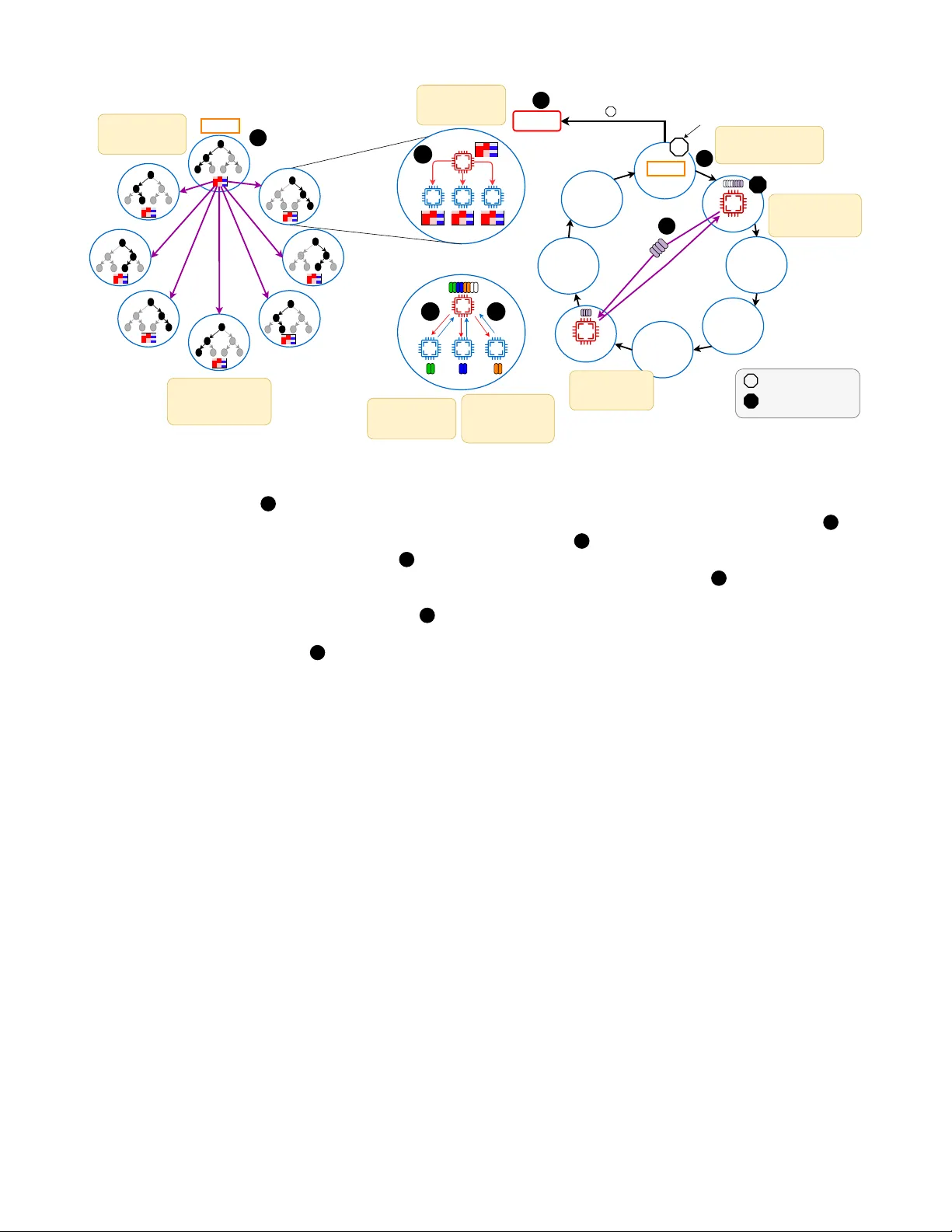
암은 단일 유전자 변이만으로는 발생하지 않으며, 여러 유전자의 변이가 동시에 존재할 때 종양이 형성된다는 ‘다중 히트’ 가설이 널리 받아들여지고 있다. 이러한 배경에서 연구자들은 암 환자와 정상 조직의 변이 매트릭스를 이용해 어떤 유전자 조합이 암을 가장 잘 설명하는지를 찾고자 한다. 전통적인 접근법은 이 문제를 가중 집합 커버(Weighted Set Cover, WSC) 문제로 모델링한다. 여기서는 모든 가능한 h‑gene 조합을 열거하고, 각 조합이 암 샘플을 얼마나 많이 커버하고 정상 샘플을 얼마나 적게 포함하는지를 점수화한다. 그러나 인간 유전체의 유전자 수가 약 20,000개이므로, h가 4가 되면 조합 수는 10^15 수준으로 폭발적으로 증가한다. 이 때문에 기존의 완전 탐색 방식은 실용적이지 않으며, GPU 가속이나 제한된 히트 수(2~3)만을 다룰 수 있었다.
본 논문은 이러한 한계를 극복하기 위해 ‘Pruned Depth‑First Search (P‑DFS)’라는 새로운 알고리즘을 제안한다. 핵심 아이디어는 암 변이 매트릭스가 매우 희소하다는 사실을 활용해 탐색 공간을 사전에 크게 축소하는 것이다. 구체적으로, 각 유전자와 샘플 간 변이 여부를 비트셋으로 저장하고 비트 연산을 통해 빠른 교집합 검사를 수행한다. 탐색 과정에서는 현재까지 선택된 유전자 집합이 커버하는 샘플 수가 사전에 정의된 최소 커버리지를 만족하지 못하면 해당 서브트리를 즉시 차단한다. 이는 ‘반단조성(anti‑monotonicity)’ 원리를 적용한 것으로, 불필요한 조합을 조기에 배제함으로써 전체 탐색 노드 수를 2 % 이하로 감소시킨다.
프루닝이 끝난 후보 집합에 대해서는 기존의 그리디 WSC 알고리즘을 그대로 적용한다. 각 조합 γ에 대해 가중치 F(γ)=α·T⁺(γ)+(1−α)·T⁻(γ) (α≈0.1) 를 계산하고, 가장 높은 점수를 가진 조합을 선택한다. 선택된 조합이 커버한 샘플을 매트릭스에서 제거하고, 이 과정을 모든 암 샘플이 커버될 때까지 반복한다.
실험은 일본 RIKEN의 Fugaku 슈퍼컴퓨터(147,456 MPI 랭크)를 이용해 수행되었다. 4‑hit, 5‑hit, 6‑hit 조합에 대해 각각 탐색된 후보 수, 프루닝 비율, 실행 시간, 그리고 최종 커버리지를 측정하였다. 결과는 4‑hit 경우 전체 조합의 90 %~98 %를 프루닝으로 제외했으며, 전체 실행 시간은 기존 Exhaustive WSC 대비 평균 183배 가속되었다. 또한, 프루닝 전후의 최종 선택 조합이 암‑정상 구분 정확도에서는 유의미한 차이를 보이지 않아, 효율성을 크게 높이면서도 정확성을 유지함을 확인하였다.
알고리즘 복잡도 분석에서는 프루닝 전후의 탐색 공간을 각각 O( C(G,h) )와 O( φ·C(G,h) ) 로 표현했으며, φ는 실제 탐색된 노드 비율(≤0.02)이다. 따라서 실질적인 시간 복잡도는 거의 선형에 가깝게 감소한다. 또한, 비트셋 기반 연산과 MPI 기반 작업 스틸링 스케줄러를 결합해 노드 간 부하를 자동으로 균형 맞춤으로써 대규모 클러스터에서도 높은 확장성을 확보했다.
한계점으로는 프루닝 기준이 데이터의 희소성에 크게 의존한다는 점이다. 변이 빈도가 높은 암 유형에서는 프루닝 효율이 감소할 수 있다. 또한, 현재 구현은 h 값을 사전에 고정하고 탐색하지만, 실제 임상에서는 가변적인 히트 수가 필요할 수 있어 동적 h‑selection 메커니즘이 추가적으로 요구된다.
결론적으로, 본 연구는 희소성 기반 프루닝과 고성능 병렬 DFS를 결합해 기존 WSC 기반 다중 히트 탐색의 병목을 근본적으로 해소했으며, 대규모 암 유전체 데이터에서 실용적인 다중 변이 조합을 신속히 도출할 수 있는 새로운 알고리즘 프레임워크를 제시한다. 이는 암 발생 메커니즘 이해와 맞춤형 복합 치료제 개발에 중요한 도구가 될 것으로 기대된다.
**
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기