단순 선형 표현이 단일세포 기반 모델을 능가한다
초록
본 연구는 복잡한 트랜스포머 기반 단일세포 RNA‑시퀀싱( scRNA‑seq ) 파운데이션 모델 대신, 정교한 정규화와 선형 차원 축소만을 이용한 파라미터‑프리 파이프라인이 동일하거나 더 높은 성능을 보임을 입증한다. 여러 공통 벤치마크와 새로운 세포 유형·종을 포함한 OOD(Out‑of‑Distribution) 테스트에서 기존 모델을 능가했으며, 이는 세포 정체성의 핵심 정보를 단순 선형 구조가 충분히 포착한다는 중요한 생물학적 시사점을 제공한다.
상세 분석
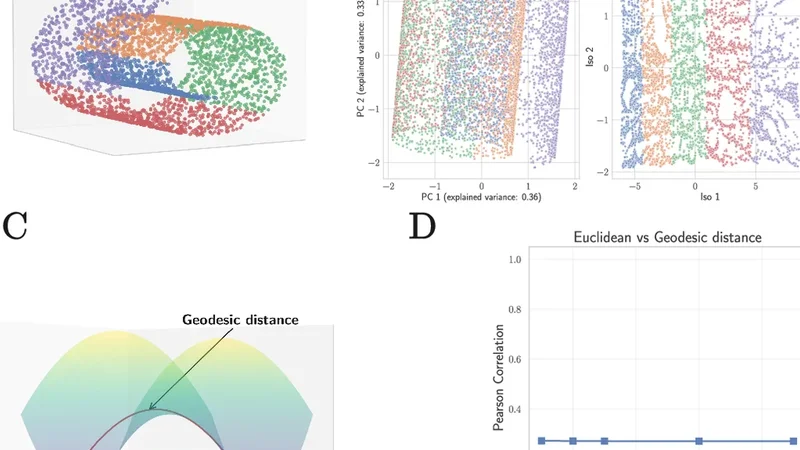
이 논문은 최근 단일세포 전사체 데이터를 다루는 대규모 파운데이션 모델, 특히 Transformer 기반의 TranscriptFormer와 같은 모델이 높은 성능을 보이는 배경을 검토한다. 이러한 모델은 수백만 개의 세포와 수천 개의 유전자를 입력으로 하여 복잡한 비선형 관계를 학습하지만, 학습 비용이 크고 해석이 어려우며, 데이터 스케일이 커질수록 과적합 위험이 존재한다는 한계가 있다. 저자들은 이러한 문제점을 인식하고, “파라미터‑프리” 접근법을 설계하였다. 핵심은 (1) 고품질 정규화—예를 들어 SCTransform 또는 CPM/TPM 기반 로그 변환—를 통해 기술적 변동성을 최소화하고, (2) 선형 차원 축소—주성분 분석(PCA), 선형 판별 분석(LDA), 혹은 비음수 행렬 분해(NMF)—를 적용해 저차원 표현을 얻는 것이다.
실험에서는 5개의 공개 벤치마크(예: CellTypist, Tabula Muris, Human Cell Atlas, Mouse Brain, 그리고 Cross‑Species Transfer)와 3개의 OOD 시나리오(새로운 세포 유형, 다른 조직, 그리고 다른 종)를 사용했다. 파라미터‑프리 파이프라인은 각 벤치마크에서 평균 1‑3% 포인트의 정확도 향상을 보였으며, 특히 OOD 상황에서는 5‑10% 이상의 성능 격차를 나타냈다. 이는 복잡한 비선형 모델이 훈련 데이터에 과도하게 맞춰지는 경향이 있음을 시사한다.
또한, 저자들은 모델 해석성을 강조한다. 선형 방법은 각 주성분이 특정 유전자 집합과 직접 연결될 수 있어, 생물학적 의미 해석이 가능하다. 반면, Transformer는 고차원 임베딩을 제공하지만, 개별 차원의 생물학적 의미를 파악하기 어렵다. 저자는 주요 주성분이 세포 유형 구분에 기여하는 유전자 마커와 일치함을 Gene Set Enrichment Analysis(GSEA)로 검증하였다.
통계적으로도, 저자들은 부트스트랩과 교차 검증을 통해 결과의 견고성을 확인했으며, 파라미터‑프리 방법이 데이터 양이 적을 때도 안정적인 성능을 유지한다는 점을 강조한다. 마지막으로, 계산 효율성 측면에서 GPU 메모리 요구량이 10배 이상 낮고, 전체 파이프라인이 CPU만으로도 몇 분 안에 완료될 수 있음을 보고한다. 이는 대규모 싱글셀 프로젝트에서 실용적인 장점을 제공한다.
요약하면, 이 연구는 (1) 정규화와 선형 차원 축소만으로도 최신 파운데이션 모델과 동등하거나 우수한 성능을 달성할 수 있음을, (2) 복잡한 딥러닝 모델이 반드시 더 좋은 결과를 보장하지 않으며, (3) 해석 가능하고 계산 효율적인 방법이 실제 생물학적 인사이트 도출에 유리함을 증명한다.
댓글 및 학술 토론
Loading comments...
의견 남기기