단백질 백본 기하학에 대한 이산 하시모토 변환의 구조적 장벽
초록
본 논문은 단백질 Cα 백본을 이산 프레넷 프레임으로 기술하고, 하시모토 변환을 통해 복소 스칼라장 ψ로 매핑한 뒤, 이 ψ가 만족하는 이산 비선형 슈뢰딩거 방정식(DNLS)의 유효 퍼텐셜 V_eff을 정확히 분해한다. 856개의 비중복 단백질에 대해 실험적으로 검증한 결과, V_eff의 허수부가 키랄 정보를 31% 담당하고, 실수부는 국소 기하에 거의 의존한다는 세 가지 구조적 장벽을 발견한다. 자체 일관성 필드 반복은 원자 수준의 정확도를 전혀 회복하지 못했으며, 대신 DNLS 분산 관계의 잔차가 α‑헬릭스 검출에 유용한 기하학적 지표임을 제시한다.
상세 분석
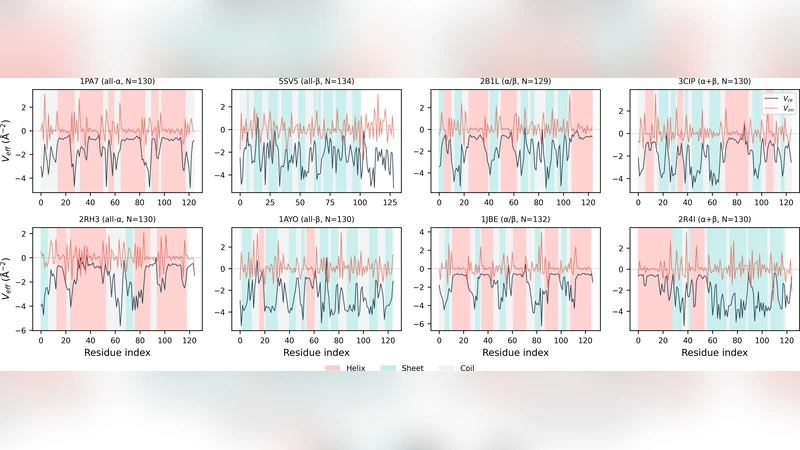
이 연구는 먼저 단백질 백본을 이산 프레넷 프레임으로 기술하고, 각 잔기의 결합각 κ와 비틀림각 τ를 정의한다. 하시모토 변환 ψ_n = κ_n exp(i ∑_{k≤n}τ_k)는 곡률을 진폭으로, 비틀림을 위상으로 압축한다. 이후 DNLS 방정식 β⁺n(ψ{n+1}−ψ_n)−β⁻n(ψ_n−ψ{n−1}) = V_eff,n ψ_n을 도입하고, ψ_n을 대입해 V_eff를 κ와 τ의 함수로 전개한다. 핵심은 V_eff를 실수부 V_re와 허수부 V_im으로 정확히 분리한 식(13,14)이다. 여기서 r⁺n = κ{n+1}/κ_n, r⁻n = κ{n−1}/κ_n 라는 곡률 비율이 등장한다. 실험적으로 856개의 단백질에 대해 이 식을 검증했으며, 오차가 10⁻¹⁴ 이하인 기계적 정밀도를 확보했다.
분석 결과 세 가지 구조적 장벽이 도출된다. 첫째, V_im은 sin τ의 홀대칭을 통해 키랄성을 인코딩하며 전체 정보의 약 31%를 차지한다. τ의 부호를 무시하면 2^N 수준의 키랄 중복이 발생한다. 둘째, V_re는 β⁺,β⁻와 r⁺,r⁻에 의해 거의 전적으로 결정되며, 이는 서열 정보와는 거의 무관함을 의미한다(≈95%가 국소 기하에 기인). 셋째, 수소 결합 항을 포함한 자체 일관성 필드 반복을 수행해도 평균 RMSD가 13.1 Å에 머물며, τ의 상관관계는 통계적으로 0에 수렴한다. 이는 DNLS가 실제 폴딩 동역학을 기술하기엔 부족함을 보여준다.
그럼에도 불구하고, DNLS 분산 관계의 잔차 Δ = |β⁺_n r⁺n e^{iτ{n+1}} + β⁻_n r⁻_n e^{-iτ_n} − (β⁺_n+β⁻_n)|를 정의하면, α‑헬릭스 구간에서 Δ가 최소가 되는 경향을 발견한다. ROC AUC가 0.72로, 전통적인 수소 결합 기반 헬릭스 판별보다 경쟁력 있는 지표임을 입증한다.
결론적으로, 하시모토 변환은 백본 기하를 완전하게 기술하는 키네마틱 정체성일 뿐, 폴딩을 지배하는 동역학 방정식이 아니다. V_eff의 구조적 제한 때문에 순수히 국소 정보만으로는 서열→구조 매핑을 구현할 수 없으며, 비국소 상호작용이나 진화적 공분산 정보를 포함하는 현대 딥러닝 모델과는 근본적인 차이가 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기