다중스케일 그래프 기반 히스토패톨로지‑공간전사체 통합 학습 SIGMMA
초록
SIGMMA는 HE 조직 이미지와 셀 수준 공간전사체 데이터를 다중 스케일로 정렬하는 새로운 프레임워크이다. 이미지에서는 다중 크롭을 이용해 마이크로·메소·매크로 레벨의 임베딩을 추출하고, 전사체는 셀 그래프를 계층적으로 확장해 각 스케일에 맞는 그래프 토폴로지를 만든다. 두 모달리티의 임베딩을 다중‑스케일 대비형 손실로 동시에 정렬함으로써, 기존 단일 스케일 방법보다 유전자 발현 예측과 교차 모달 검색에서 각각 평균 9.78 %와 26.93 %의 성능 향상을 달성한다. 또한 학습된 임베딩은 조직의 다중 조직학적 구조를 의미 있게 반영한다.
상세 분석
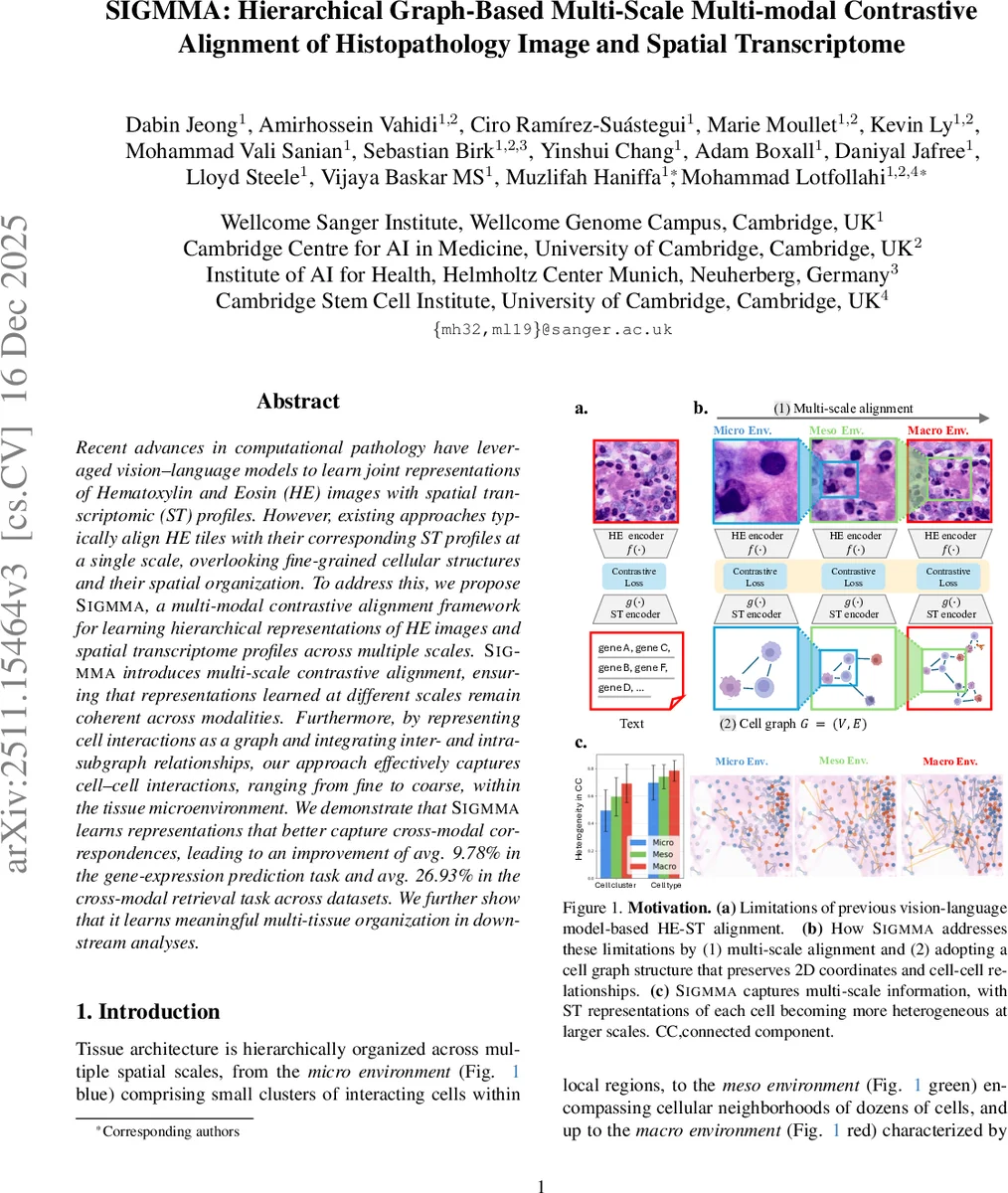
SIGMMA는 기존 비전‑언어(VL) 기반 HE‑ST 정렬이 1차원 유전자 시퀀스로 ST를 표현해 2차원 공간 정보와 세포 간 상호작용을 소실한다는 한계를 정확히 짚고 있다. 이를 해결하기 위해 저자는 세 가지 핵심 설계를 제안한다. 첫째, HE 이미지에 대해 다중‑크롭(multi‑crop) 전략을 적용해 4×4(마이크로), 2×2(메소), 1×1(매크로) 패치로 분할하고, 사전학습된 이미지 파운데이션 모델(DINO 기반)으로 각각의 패치를 임베딩한 뒤 평균 풀링해 스케일별 타일 임베딩(z_micro, z_meso, z_macro)을 만든다. 이 과정은 이미지의 계층적 조직학적 정보를 보존하면서도 파라미터 공유를 통해 효율성을 유지한다. 둘째, ST 데이터는 셀 좌표를 이용해 셀 그래프 G=(V,E)로 변환한다. 초기 노드 임베딩은 전사체 파운데이션 모델(Transformer 기반)으로 초기화하고, 스테이지별(마이크로·메소·매크로) 그래프 토폴로지를 확장한다. 확장은 ‘stochastic edge addition’ 모듈을 사용해, 현재 스케일의 이웃 패치 내에서만 새로운 엣지를 샘플링함으로써 공간 일관성을 유지한다. 각 스케일에서 GNN(메시지 패싱)과 어텐션 풀링을 거쳐 그래프 임베딩(z_S_micro, z_S_meso, z_S_macro)을 얻는다. 셋째, 다중‑스케일 대비형 손실(InfoNCE 기반)을 도입해 동일 스케일의 HE와 ST 임베딩을 서로 끌어당기고, 다른 스케일·다른 샘플 간에는 멀어지게 만든다. 이때 각 스케일별 손실을 가중합해 전체 목표함수를 구성함으로써, 미세 구조와 거시 구조 모두에서 모달리티 간 일관성을 강제한다. 실험에서는 5개의 공개 ST‑HE 데이터셋(주로 Xenium)에서 gene‑expression prediction(예: R², MAE)과 cross‑modal retrieval(mAP)에서 평균 9.78 %와 26.93 %의 개선을 보고했으며, t‑SNE·UMAP 시각화와 클러스터 분석을 통해 학습된 임베딩이 조직 내 림프구 집단, 종양 미세환경 등 의미 있는 조직학적 구역을 구분함을 확인했다. 또한, 엣지 추가 확률을 해석함으로써 특정 세포군 간 상호작용이 강조되는 것을 확인, 모델의 해석 가능성을 높였다. 전반적으로 SIGMMA는 (1) 공간 토폴로지를 보존한 그래프 기반 ST 표현, (2) 이미지와 전사체의 계층적 스케일을 일치시키는 다중‑크롭·다중‑그래프 설계, (3) 스케일 일관성을 유지하는 대비형 학습이라는 세 축을 통해 기존 VL‑기반 접근법의 한계를 뛰어넘는 통합 프레임워크를 제공한다.
댓글 및 학술 토론
Loading comments...
의견 남기기