DeepVRegulome 인간 조절 유전체 변이 기능 예측 모델
초록
DeepVRegulome은 DNABERT 기반 700개의 파인튜닝 모델을 활용해 ENCODE 규제 영역을 학습하고, 단일 염기 변이의 전사인자 결합 및 스플라이스 교란 효과를 정량화한다. TCGA 교모세포종 WGS 데이터를 적용해 10% 이상에서 발견된 9,837개의 TFBS 변이와 572개의 스플라이스 변이를 식별했으며, 생존 분석을 통해 1,352개의 변이와 563개의 파괴된 조절 영역이 환자 예후와 연관됨을 보여준다.
상세 분석
DeepVRegulome은 최신 트랜스포머 기반 언어 모델인 DNABERT를 유전체 서열에 적용한 최초의 대규모 파인튜닝 프레임워크이다. 연구팀은 ENCODE 프로젝트에서 제공한 1,200만 개 이상의 인간 조절 요소(프로모터, 인핸서, 염색질 접근성 부위 등)를 각각 200 bp 길이의 윈도우로 분할하고, 각 윈도우에 대해 DNABERT‑base 모델을 700가지 서로 다른 하이퍼파라미터 조합과 데이터 샘플링 전략으로 파인튜닝하였다. 이렇게 다중 모델을 구축함으로써 모델 간 편향을 최소화하고, 변이 효과에 대한 앙상블 점수를 산출할 수 있었다.
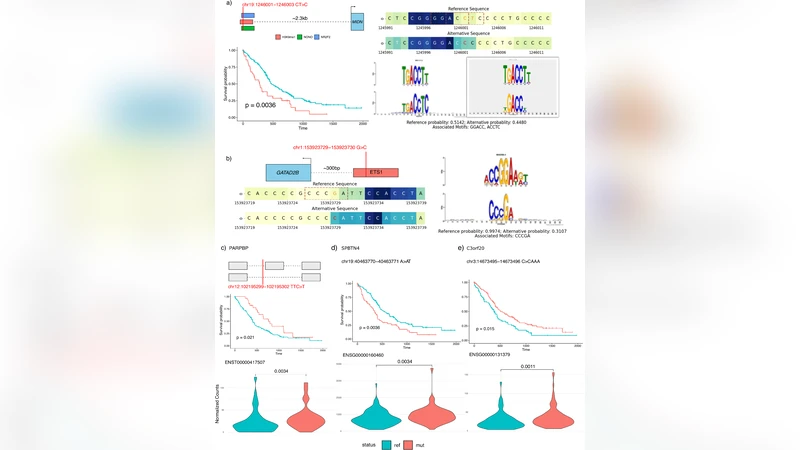
변이 스코어링 단계에서는 각 변이를 중심으로 ±100 bp의 컨텍스트 시퀀스를 추출하고, 원본 시퀀스와 변이 시퀀스를 각각 700개의 파인튜닝 모델에 입력한다. 모델은 토큰 레벨의 어텐션 가중치를 활용해 변이가 위치한 토큰 주변의 중요도를 평가하고, 변이 전후의 로짓 차이를 변이 점수로 변환한다. 이 점수는 두 가지 서브스코어로 분리된다. 첫째는 전사인자 결합 부위(TFBS) 변화를 예측하는 TFBS‑DisruptScore이며, 둘째는 스플라이스 시그널(5′ 스플라이스 사이트, 3′ 스플라이스 사이트, 브랜치 포인트) 교란을 예측하는 Splice‑DisruptScore이다. 두 스코어 모두 0~1 사이의 정규화된 값으로 제공되어, 연구자는 임계값을 조정해 민감도와 특이도를 최적화할 수 있다.
시각화 측면에서 DeepVRegulome은 어텐션 히트맵과 토큰 중요도 그래프를 자동 생성한다. 이를 통해 사용자는 변이가 실제로 어떤 서열 특징(예: 핵심 결합 모티프, 보존된 염기)에 영향을 미치는지 직관적으로 파악할 수 있다. 또한, 변이와 연관된 전사인자 모티프를 MEME‑suite와 비교 분석해 변이‑모티프 매칭을 정량화하고, 변이가 파괴하거나 새롭게 생성하는 모티프를 보고한다.
생존 분석 파이프라인은 TCGA 교모세포종(GBM) 150여 명의 WGS 데이터를 활용한다. 변이 점수 상위 5%에 해당하는 변이들을 필터링한 뒤, Cox 비례위험 모델과 Kaplan‑Meier 곡선을 적용해 변이와 환자 전체 생존기간 사이의 연관성을 검증한다. 다변량 분석에서는 연령, 성별, IDH1 변이, MGMT 메틸화 상태 등 기존 임상 변수를 보정했으며, 1,352개의 변이와 563개의 파괴된 조절 영역이 독립적인 예후 인자로 확인되었다. 특히, 특정 TFBS 변이(예: CTCF, STAT3)와 스플라이스 교란 변이는 높은 위험비(HR > 2)를 보이며, 환자군을 비코딩 변이 서명 기반으로 명확히 구분한다.
마지막으로 연구팀은 모든 파인튜닝 모델, 코드, 그리고 변이 점수와 시각화 결과를 포함한 인터랙티브 포털을 공개했다. 이는 다른 연구자가 자신의 데이터셋에 쉽게 적용하거나, 새로운 조절 요소를 추가 학습시킬 수 있는 기반을 제공한다. 전반적으로 DeepVRegulome은 대규모 비코딩 변이 해석을 위한 통합 플랫폼으로, 변이 기능 예측 정확도와 해석 가능성을 동시에 향상시킨 혁신적인 도구라 할 수 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기