파이로시퀀싱 양성 신호 수의 확률분포와 평균‑분산 특성
본 논문은 파이로시퀀싱에서 흐름 사이클 수와 염기 출현 확률에 따라 pyrogram에 나타나는 양성 신호(런)의 개수가 어떻게 분포하는지를 수학적으로 규명한다. 고정 흐름 사이클 모델과 고정 양성 신호 모델을 각각 정의하고, 생성함수(GF)를 이용해 정확한 확률식과 평균·분산을 도출하였다. 핵심 결과는 평균 양성 신호 수가 흐름 사이클 수의 약 2배에 수렴한다는 점이며, 이는 염기 확률에 무관하게 성립한다.
저자: Yong Kong
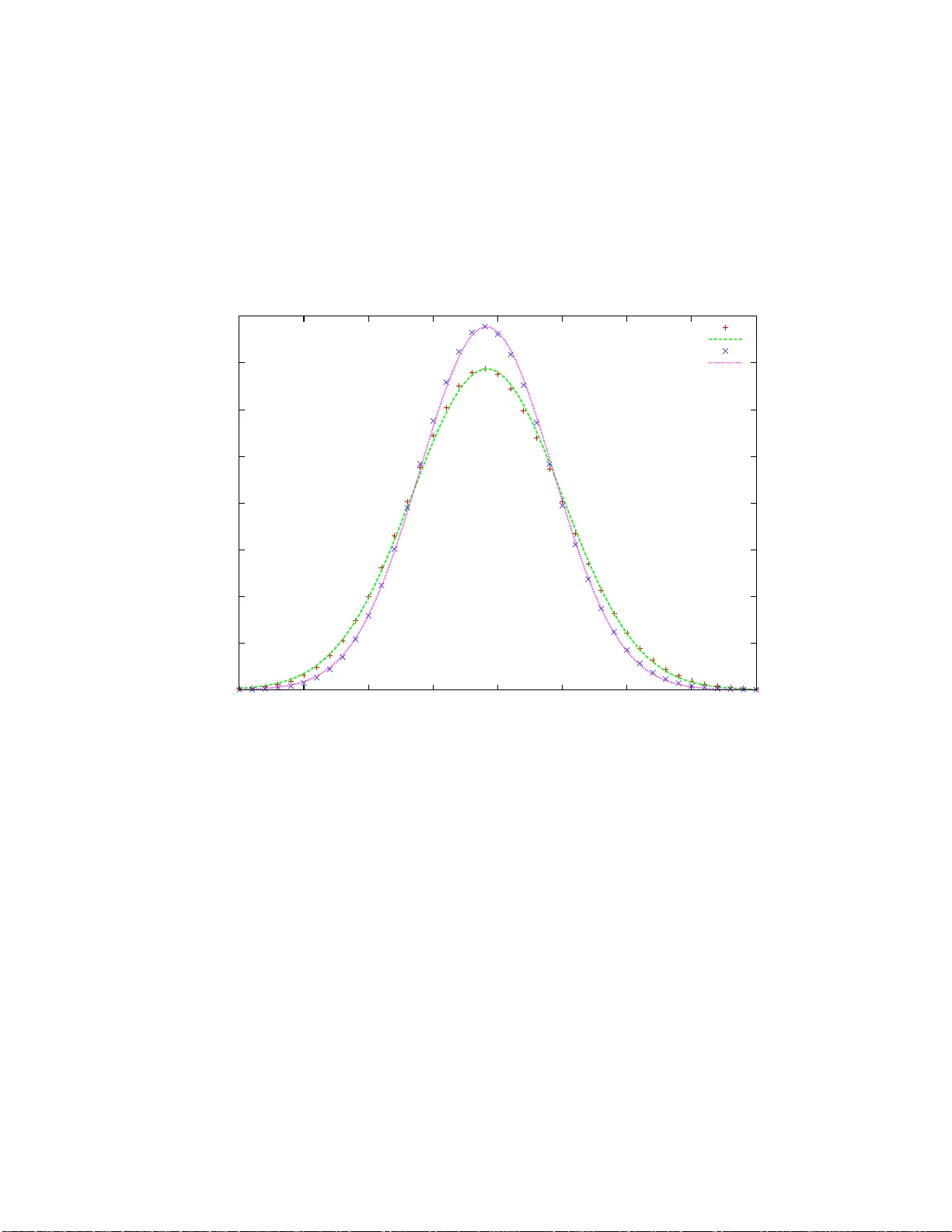
본 논문은 차세대 시퀀싱 기술 중 하나인 파이로시퀀싱(pyrosequencing)의 핵심 데이터 형태인 pyrogram에서 나타나는 양성 신호(positive signals)의 개수가 어떻게 분포하는지를 이론적으로 분석한다. 파이로시퀀싱은 네 종류의 dNTP(A, C, G, T)를 미리 정해진 순서(a‑b‑c‑d)로 순차적으로 흐르게 하여, 템플릿 DNA와 상보적인 경우에만 빛 신호가 발생한다. 양성 신호는 실제 염기 삽입을 의미하고, 0 신호는 배경 잡음에 해당한다. 양성 신호를 연속된 동일 염기의 ‘런(run)’으로 압축하면 원본 서열의 r‑seq(런 서열)를 얻으며, r‑seq의 길이 r가 바로 pyrogram에서 관측되는 양성 신호 수와 동일하다.
연구는 두 가지 확률 모델을 설정한다. 첫 번째는 ‘고정 r‑seq 길이 모델(Fixed r‑seq Length Model, FRLM)’으로, 양성 신호 수 r를 미리 정하고 흐름 사이클 f가 랜덤하게 결정되는 상황을 다룬다. 여기서 L_i(f,r) (i∈{a,b,c,d})를 “r‑seq가 길이 r이며 마지막 염기가 i이고, r번째 런이 흐름 사이클 f에서 합성된 경우의 비정규화 확률”로 정의한다. 네 개의 재귀식(식 6)은 각 염기의 이전 상태와 µ_i = p_i/(1‑p_i) (p_i는 염기 i의 출현 확률) 를 이용해 L_i를 전이시킨다. 직접적인 해는 존재하지 않지만, 생성함수 G_i(x,y)=∑_{f,r}L_i(f,r)x^f y^r 를 도입하면 폐쇄형 해를 얻는다. G_i는 µ_i와 대칭 함수 S_k(µ) (k=1~4) 로 표현되며, 네 개를 합산한 전체 생성함수 G(x,y)=G_a+G_b+G_c+G_d (식 9)는 µ_i에 대해 완전 대칭성을 가진다.
정규화는 필수적이다. L_i(f,r) 자체는 확률이 아니므로, x=1 혹은 y=1을 대입해 각각 u(r)=∑_f L(f,r)와 v(f)=∑_r L(f,r) 를 구한다. u(r)와 v(f)는 각각 r 고정, f 고정 상황에서 전체 확률 질량을 제공한다. 논문은 u(r)≈½e², v(f)≈e² (e₁=1) 라는 근사식을 제시한다. 균등 염기 확률(p_a=p_b=p_c=p_d=¼)일 때는 정확하고, 비균등일 경우에도 r·f가 충분히 크면 오차가 무시될 정도로 수렴한다.
두 번째 모델은 ‘고정 흐름 사이클 모델(Fixed Flow Cycle Model, FFCM)’이다. 여기서는 흐름 사이클 수 f를 고정하고 r을 랜덤 변수로 본다. FRLM에서 얻은 정규화된 분포를 이용해, r의 평균과 분산을 f에 대한 함수로 변환한다. 핵심 결과는 평균 r̄ ≈ 2f (식 17a, 23a) 로, 흐름 사이클당 평균 두 개의 양성 신호가 발생한다는 직관적이면서도 일반적인 사실이다. 이는 염기 출현 확률 p_i와 무관하게 성립한다. 분산 역시 선형적으로 f에 비례하지만, FRLM과 FFCM 사이에 비전(variance non‑transitivity)이 존재함을 논문은 강조한다.
통계적 특성은 모두 가우시안 근사로 수렴한다. 즉, f가 커질수록 r의 분포는 평균 2f, 분산 σ²·f (σ²는 염기 확률에 따라 달라짐) 를 갖는 정규분포에 가까워진다. 이는 중심극한정리와 일치하며, 논문은 시뮬레이션을 통해 이론적 분포와 실험적 결과가 거의 일치함을 확인한다.
실용적 의미는 크게 두 가지로 정리된다. 첫째, 베이스콜러 알고리즘에 새로운 기준을 제공한다. 기존 콜러는 고정 임계값을 사용해 양성/음성 신호를 구분하지만, 이론적 r 분포를 활용하면 현재 관측된 양성 신호 수와 기대값 사이의 차이를 최소화하도록 임계값을 동적으로 조정하는 부트스트랩 방식이 가능해진다. 이는 삽입·삭제 오류를 감소시킬 수 있다. 둘째, 실제 데이터에서 r 분포가 이론과 크게 벗어난다면 염기 간 상관관계(예: GC‑bias, 구조적 변이) 존재를 시사하는 검정 도구로 활용될 수 있다.
결론적으로, 논문은 파이로시퀀싱 데이터의 통계적 구조를 정확히 규명하고, 생성함수와 정규화 기법을 통해 평균·분산을 포함한 전체 확률분포를 제공한다. 이러한 이론적 프레임워크는 시퀀싱 장비 설계, 소프트웨어 최적화, 오류 모델링 및 데이터 품질 평가 등에 직접 적용 가능하며, 차세대 시퀀싱 기술 전반에 걸친 통계적 분석의 토대를 마련한다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기