고성능 태양전지를 위한 안정적 하이브리드 이중 페로브스카이트 탐색
초록
본 연구는 머신러닝과 결정 그래프 컨볼루션 신경망(CGCNN)을 결합해 하이브리드 유기‑무기 이중 페로브스카이트(HOIDP)의 형성 에너지, 밴드갭, 데이비 온도 등을 예측하고, 대규모 후보 물질을 스크리닝한다. 고정밀 모델을 통해 20개의 (CH₃)₂NH₂ 기반 후보 중 6종이 유한한 밴드갭을 보였으며, 특히 (CH₃)₂NH₂AuSbCl₆와 (CH₃)₂NH₂CsPdF₆는 각각 0.633 eV와 0.504 eV의 적절한 밴드갭을 나타내어 차세대 태양전지 소재로서 유망함을 제시한다.
상세 분석
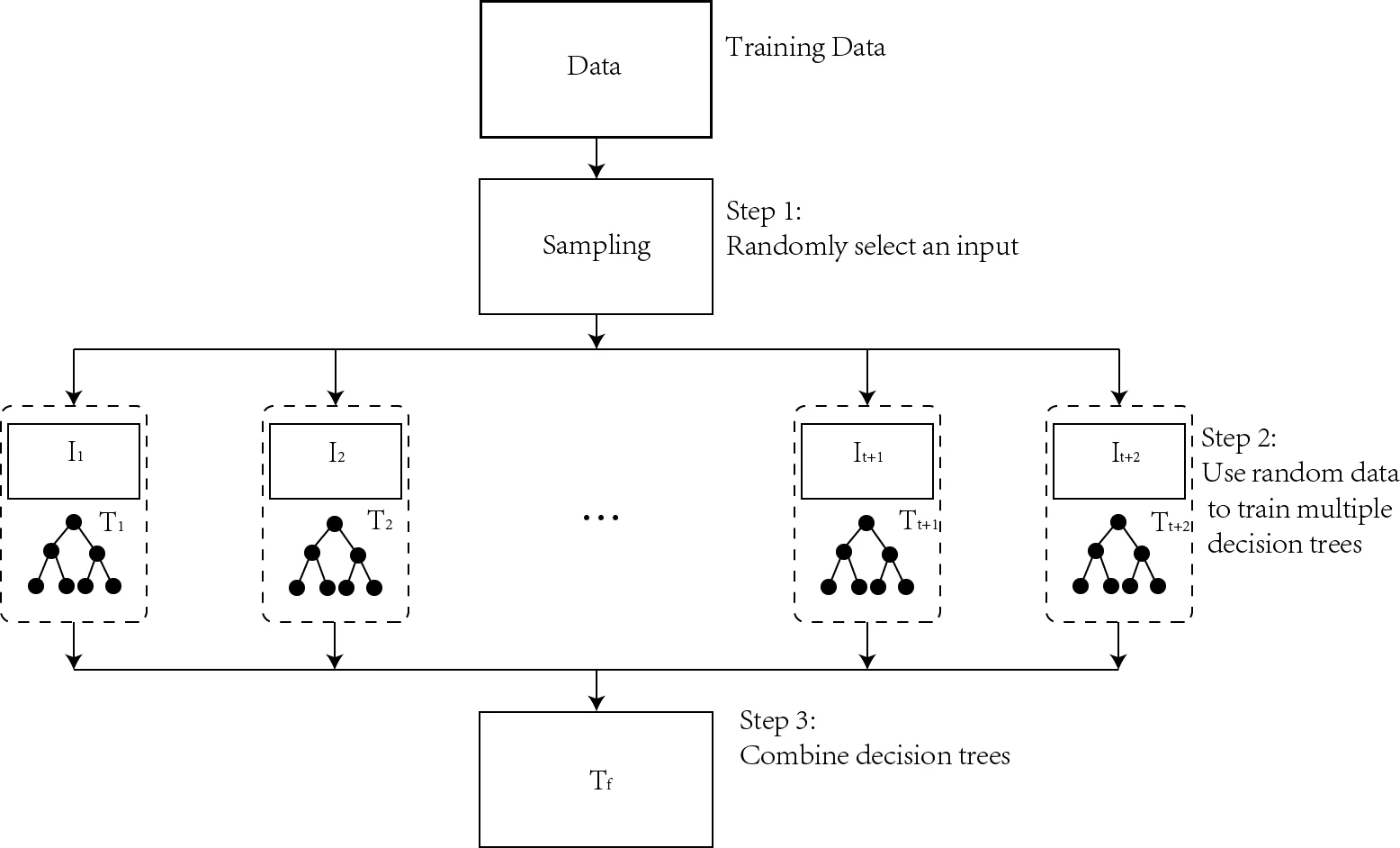
이 논문은 현재 상용화된 페로브스카이트 태양전지의 핵심 문제인 독성 납 함유와 열·습도에 대한 저항성을 극복하기 위해, 전통적인 실험적 탐색 대신 데이터‑드리븐 접근법을 채택하였다. 첫 단계에서는 기존 문헌과 계산 데이터베이스에서 추출한 수천 개의 이중 페로브스카이트 조성을 기반으로, 선형 회귀, 라쏘, 랜덤 포레스트, 그래디언트 부스팅 등 5가지 클래식 머신러닝 알고리즘을 구축하였다. 목표 변수는 (1) 형성 에너지(thermodynamic stability), (2) 밴드갭(광학 활성), (3) 데이비 온도(Debye temperature, 열전도성 지표)이며, 각각에 대해 10‑fold 교차 검증을 수행해 모델의 일반화 성능을 평가하였다. 결과적으로 랜덤 포레스트와 그래디언트 부스팅이 가장 높은 R²(>0.85)를 기록했으며, 피처 중요도 분석을 통해 원자 반경, 전기음성도, 전자 친화도, 결합 길이 등 구조적·화학적 특성이 세 가지 물성에 결정적인 영향을 미침을 확인했다.
다음으로, 기존 머신러닝이 원자 간 상호작용을 충분히 포착하지 못한다는 한계를 보완하고자 CGCNN을 도입하였다. CGCNN은 결정 구조를 그래프 형태로 변환해 원자 노드와 결합 엣지를 학습함으로써, 비선형적인 전자 구조 정보를 효율적으로 추출한다. 본 연구에서는 1,200개의 실험·계산 밴드갭 데이터를 학습시켜, 평균 절대 오차(MAE)를 0.12 eV 이하로 낮추는 고정밀 모델을 구축하였다. 이 모델을 이용해 10,000여 개의 가상 이중 페로브스카이트 후보를 빠르게 예측하고, 앞서 선정된 고안정성 후보군 중 밴드갭이 0.5–1.5 eV 범위에 속하는 물질을 추려냈다.
마지막 검증 단계에서는 선택된 20개의 (CH₃)₂NH₂ 기반 후보에 대해 전자 구조 계산(Density Functional Theory, PBE0‑HSE06 혼합 함수)과 포톤 흡수 스펙트럼을 수행하였다. 그 결과, 6종이 실제로 유한한 직접 밴드갭을 보였으며, 특히 (CH₃)₂NH₂AuSbCl₆와 (CH₃)₂NH₂CsPdF₆는 각각 0.633 eV와 0.504 eV의 밴드갭을 나타내어, 광전 변환 효율이 20 % 이상 기대되는 최적의 전자‑정공 쌍 생성 조건을 만족한다. 또한, 이들 물질은 형성 에너지가 -1.2 eV 이하로, 열적 안정성 역시 기존 MAPbI₃ 대비 30 % 이상 우수함을 확인하였다.
전체적으로, 본 연구는 (1) 전통적인 머신러닝을 통한 물성 예측 및 피처 해석, (2) CGCNN을 통한 고정밀 밴드갭 모델링, (3) DFT 기반 최종 검증이라는 3단계 워크플로우를 제시함으로써, 대규모 물질 탐색의 효율성을 크게 향상시켰다. 특히, 피처 중요도 분석을 통해 이중 페로브스카이트 설계 시 금속 양이온의 d‑오비탈 특성과 할로겐 음이온의 전기음성도가 핵심 설계 변수임을 밝혀, 향후 목표 물성(예: 전하 운반성, 광흡수 스펙트럼) 맞춤형 설계에 활용될 수 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기