고차원 유전체 데이터의 가우시안 그래프 모델 비교 연구
초록
본 논문은 유전체 고처리량 데이터에서 부분 상관을 추정하기 위한 세 가지 방법—Moore‑Penrose 의사역행렬(PINV), 잔차 상관법(RCM), ℓ₂ 정규화 공분산 방법(ℓ₂C)—을 시뮬레이션과 실제 Arabidopsis 이소프레노이드 경로 데이터에 적용해 성능과 계산 속도를 비교한다. 결과는 PINV가 변수 수가 변할 때 AUC가 불안정한 반면, ℓ₂C와 RCM은 높은 정확도를 보이며 ℓ₂C가 RCM보다 현저히 빠르다는 것이다. ℓ₂C를 이용해 두 경로 간 전사 네트워크를 재구성하고, HMGS와 HDS가 교차 조절에 핵심 역할을 함을 제시한다.
상세 분석
이 연구는 n < p 상황, 즉 샘플 수보다 변수(유전자) 수가 훨씬 많은 전형적인 유전체 데이터에 초점을 맞춘다. 전통적인 GGM에서는 공분산 행렬 Σ를 직접 역행렬화해 정밀도 행렬 Θ = Σ⁻¹을 얻고, Θ의 비대각 원소를 이용해 부분 상관 ρ_{ij·V{i,j}} = ‑θ_{ij}/√(θ_{ii}θ_{jj})를 계산한다. 그러나 n < p이면 Σ는 특이행렬이 되어 역행렬이 존재하지 않으므로 정규화가 필수적이다.
첫 번째 방법인 PINV는 특이값 분해(SVD)를 이용해 Σ의 의사역행렬 Σ⁺를 구한다. 이때 작은 특이값을 0으로 처리하거나 역수를 취함으로써 수치적 불안정성이 발생한다. 논문에서는 시뮬레이션 결과, p가 n에 근접할 때(특히 p≈n ≈ 200) AUC가 급격히 변동하며 “공명 효과”라 부르는 현상이 나타난다. 이는 작은 특이값이 크게 증폭돼 부분 상관 추정에 잡음이 섞이기 때문이다.
두 번째 방법인 RCM은 각 변수 쌍(i, j)에 대해 나머지 p‑2 변수를 고정하고 정규화 최소제곱(Ridge) 회귀를 수행한다. 회귀계수 β^{(i)}와 β^{(j)}를 구한 뒤, 잔차 r_i, r_j를 계산하고 이들의 피어슨 상관을 부분 상관으로 정의한다. λ는 Leave‑One‑Out 교차검증으로 최적화한다. 이 접근법은 변수 간 직접적인 조건부 독립성을 평가하므로 이론적으로 정확하지만, 모든 (i, j) 쌍에 대해 회귀를 수행해야 하므로 계산 복잡도가 O(p² · n · p) 정도로 급격히 증가한다. 실험에서는 RCM이 ℓ₂C에 비해 10배 이상 오래 걸리는 것으로 보고되었다.
세 번째 방법인 ℓ₂C는 로그우도에 L2 페널티 λ‖Θ‖_F²를 추가한 정규화 문제를 풀어 Θ를 직접 추정한다. 최적화 조건은 Θ⁻¹ − 2λΘ = S이며, 이는 고유값 문제로 변환된다. Σ의 고유값 s_i에 대해 θ_i = (‑s_i + √(s_i² + 8λ))/4λ 로 닫힌 형태 해를 얻는다. 따라서 전체 행렬을 고유분해 한 번 수행하면 정밀도 행렬을 즉시 계산할 수 있어 시간 복잡도가 O(p³) (고유분해 비용) 수준이다. λ는 20번의 랜덤 트레인/밸리데이션 분할을 통해 로그우도 검증으로 선택한다.
시뮬레이션에서는 세 가지 네트워크 토폴로지(무작위, 허브, 클리크)를 각각 p = 50, 200, 400, n = 20, 200, 500 조합으로 생성했다. n > p인 경우 세 방법 모두 AUC≈1로 동일한 성능을 보였지만, n < p에서는 PINV가 불안정해졌다. ℓ₂C와 RCM은 AUC가 0.95 이상으로 일관되었으며, 특히 무작위 토폴로지에서 n = 20, p = 200/400일 때 RCM이 ℓ₂C보다 약 10 % 높은 AUC를 기록했다. 그러나 실행 시간은 ℓ₂C가 0.03 s 수준에 머무는 반면, RCM은 0.8 s~1.0 s 정도로 현저히 느렸다.
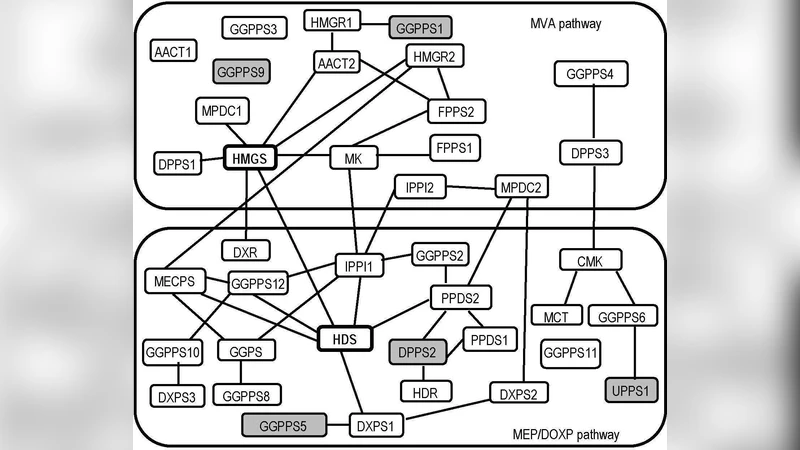
실제 데이터 적용에서는 Arabidopsis thaliana의 이소프레노이드 경로(세포질 MV A와 엽록체 MEP/DOXP)와 56개의 다른 대사 경로에서 총 834개의 유전자 발현 데이터를 사용했다. ℓ₂C를 적용해 95 % 부트스트랩 신뢰구간을 구하고, 0을 포함하지 않는 경우에만 엣지를 인정했다. 결과 네트워크는 44개의 엣지를 포함했으며, 각 경로 내부에 고도로 연결된 모듈이 형성되었다. 특히 HMGS와 HDS가 두 경로 사이에 교차 조절 역할을 하는 후보 유전자로 도출되었다. 이는 기존 연구에서 보고된 대사적 교차와 일치하며, 전사 수준에서도 상호작용이 존재함을 시사한다.
전체적으로 ℓ₂C는 정확도와 계산 효율성 측면에서 실용적인 선택이며, 대규모 유전체 네트워크 추정에 적합함을 입증한다. RCM은 이론적으로는 강력하지만, 고차원 데이터에서는 계산 비용이 제한 요인이다. PINV는 간단하지만 특이값 처리에 민감해 n≈p 상황에서 신중히 사용해야 한다.
댓글 및 학술 토론
Loading comments...
의견 남기기