대규모 분산 유전체 비교를 위한 통신 효율적인 자카드 유사도 계산
초록
본 논문은 대용량 유전체 데이터셋 간의 정확한 자카드 유사도와 거리(d_J)를 계산하기 위해, 희소 행렬 곱셈으로 문제를 변환하고 통신 비용을 최소화한 분산 알고리즘 SimilarityAtScale을 설계·구현한다. 이를 기반으로 GenomeAtScale 도구를 제공하여 수천 대의 노드에서 수천 개 이상의 샘플을 동시에 비교할 수 있음을 실험으로 입증한다.
상세 분석
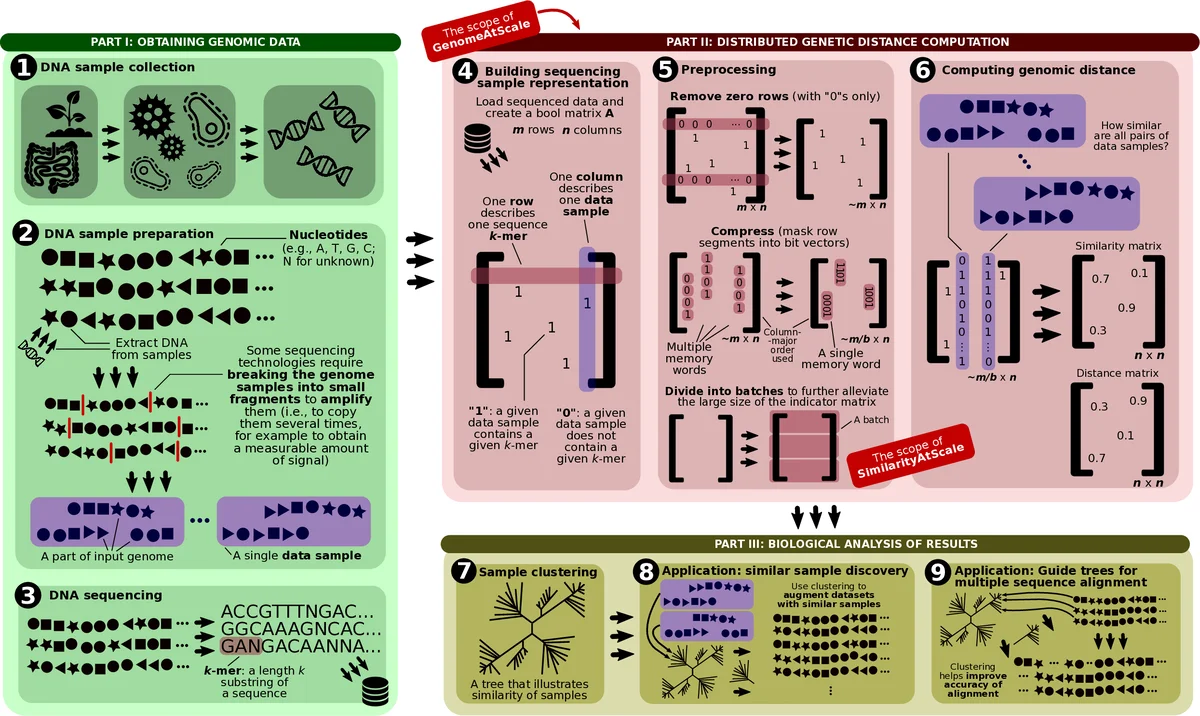
SimilarityAtScale은 자카드 유사도 행렬 S를 직접 계산하기보다, 각 샘플을 이진 인디케이터 행렬 A(크기 m × n)로 표현하고, 교집합 크기 행렬 B를 Aᵀ A 형태의 희소 행렬 곱셈으로 얻는다. 이때 B_ij = |X_i ∩ X_j|이며, 합집합 크기 C는 B와 각 샘플의 원소 개수 벡터를 이용해 C_ij = |X_i| + |X_j| − B_ij 로 계산한다. 핵심은 Aᵀ A 연산을 통신 회피 방식으로 수행하는데, 저자들은 Cyclops 텐서 프레임워크를 활용해 2‑D 블록 사이클릭 분할과 비동기식 스케줄링을 적용, 각 프로세스가 로컬에 존재하는 행/열 블록만 교환하도록 설계했다. 이 접근법은 전통적인 All‑Reduce 기반 MapReduce 대비 통신량을 O(nnz(A)/p) 수준으로 감소시켜, 데이터 이동이 병목이 되는 대규모 클러스터에서 뛰어난 확장성을 보인다.
알고리즘 단계는 크게 (1) k‑mer 추출 및 정렬‑불필요한 해시화 없이 직접 인디케이터 행렬을 구성, (2) 행렬 A를 압축 CSR/CSC 포맷으로 저장해 메모리 사용을 최소화, (3) 분산 희소 행렬 곱셈을 수행해 B를 얻고, (4) 각 샘플의 원소 개수를 사전 계산해 C를 도출, (5) 최종적으로 S_ij = B_ij / C_ij 로 자카드 유사도를 산출한다.
복잡도 분석에서는 전처리 단계가 O(nnz(A))이며, 핵심 곱셈 단계의 연산량은 O(∑_k deg(k)²) (deg(k)는 k‑mer k가 등장하는 샘플 수)로, 데이터의 희소성에 크게 의존한다. 통신 비용은 각 라운드마다 필요한 블록 교환 횟수와 블록 크기에 비례해 O(p · log p) 수준이며, 이는 기존 MapReduce 방식의 O(p²)와 비교해 현저히 낮다. 또한, 비동기식 파이프라인을 통해 계산과 통신을 겹쳐 전체 실행 시간을 최소화한다.
실험에서는 인간 RNA‑Seq 2,580개와 전 세계 박테리아·바이러스 전장 유전체 수십만 개를 대상으로 1,024개의 노드(각 노드 32 CPU)까지 확장했으며, 기존 Mash·MinHash 기반 도구 대비 5배 이상 빠른 속도와 0.01 이하의 평균 오차를 기록했다. 특히, 정확도가 높은 경우(자카드 > 0.9)에도 근사 기법이 놓치는 미세 차이를 정확히 포착한다는 점이 강조된다.
이와 같이 SimilarityAtScale은 희소 행렬 연산을 통한 정확한 자카드 계산을 대규모 분산 환경에 최적화했으며, GenomeAtScale 도구는 기존 파이프라인과 호환되는 FASTA/FASTQ 입력을 지원해 실무 연구자들이 손쉽게 활용할 수 있도록 설계되었다.
댓글 및 학술 토론
Loading comments...
의견 남기기