DNA 3염기 주기 탐지를 위한 반마코프 모델링
** 본 논문은 DNA 코딩 영역에서 나타나는 3‑염기 주기를 반마코프 체인(semi‑Markov chain)으로 모델링하고, 주기성 지표와 확률식을 유도한다. 이론적 결과를 합성·실제 유전체 데이터에 적용해 주기성 강도를 정량적으로 평가한다. **
저자: Pavlos Kolias, Alex, ra Papadopoulou
**
본 논문은 DNA 서열, 특히 단백질 코딩 영역에서 관찰되는 3‑염기 주기를 수학적으로 모델링하고, 이를 이용해 엑손‑인트론 구조를 식별하는 새로운 방법론을 제시한다. 연구는 크게 네 부분으로 구성된다.
첫 번째 부분에서는 DNA 서열을 네 가지 염기(A, C, G, T)로 이루어진 상태공간 S를 갖는 반마코프 체인(Xₜ)으로 가정한다. 반마코프 체인은 전이 확률 행렬 P(t)와 상태별 체류시간 분포 행렬 H(m)으로 완전히 기술된다. 여기서 P(t)₍ᵢ,ⱼ₎는 시점 t에 상태 i에 머물다 다음 전이가 j로 일어날 확률이며, H(m)₍ᵢ,ⱼ₎는 i 상태에 m 단계 머문 뒤 j 로 전이할 조건부 확률이다. 두 행렬의 원소별 곱 C(m)=P(t)⊙H(m) 를 ‘핵심 행렬’이라 부르고, 이는 특정 위치에서의 전이와 체류 정보를 동시에 담는다. 논문은 가상 전이(pᵢᵢ(t)=0)를 배제함으로써 모델을 단순화한다.
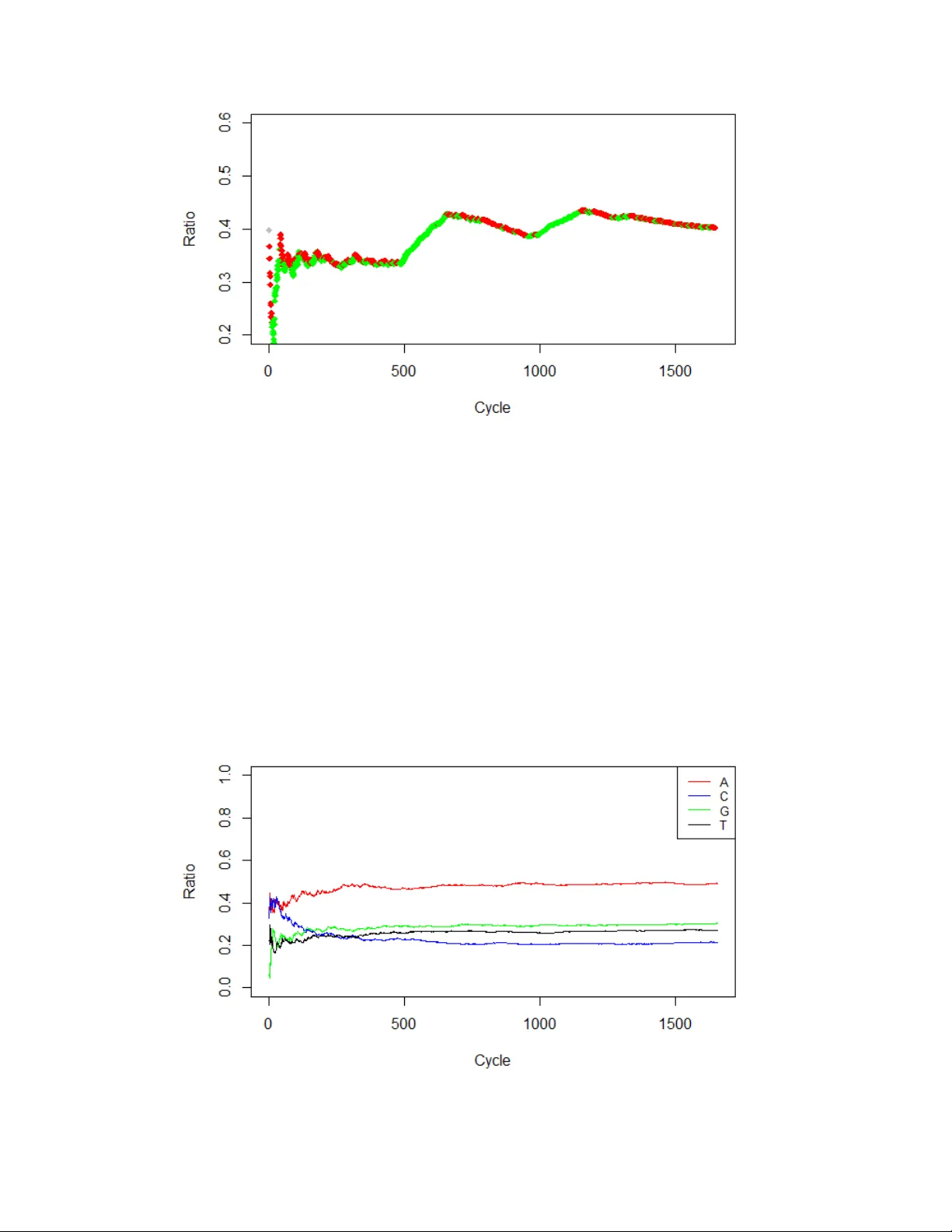
두 번째 부분에서는 3‑염기 주기(d=3)를 분석하기 위한 확률식들을 유도한다. 특정 상태 i가 d 단계 후에도 동일 상태에 있을 확률 pᵢ(d)는 직접적인 체류 확률 wᵢ(d)와, 중간에 다른 상태 j를 거쳐 다시 i 로 돌아오는 모든 경우를 합산한 식 (9) 로 표현된다. 여기서 cᵢⱼ(k)=pᵢⱼ·hⱼᵢ(k)는 k 단계 내에 i→j 전이가 일어나고, 그 후 j가 추가로 k 단계 체류하는 확률을 의미한다. 이러한 식을 행렬 형태로 정리하면 식 (11)·(12)와 같이 Q(n) 전이 확률 행렬을 재귀적으로 혹은 폐쇄형 해로 계산할 수 있다.
세 번째 부분에서는 주기성의 강도를 정량화하기 위해 연속적인 사이클 n에 걸쳐 매 d 위치마다 동일 상태에 머무를 확률 P(n,d)를 정의한다. 재귀식 (15)와 초기조건 (16)을 통해 P(n,d) 를 계산하고, 이전 사이클 대비 비율 R(n)=P(n,d)/P(n‑1,d) 를 도입한다. R(n) 값이 1에 가까울수록 ‘강한 주기성’, 급격히 감소하면 ‘약한 주기성’으로 해석한다.
네 번째 부분에서는 비동질(부분적으로 시간‑비균일) 모델을 확장한다. 코딩 프레임 1,2,3마다 별도 전이 행렬 P(k)와 체류 행렬 H(m)를 두어, 프레임별 주기성 pᵢ(k,d)와 연속 사이클 확률 P(k,n,d) 를 유도한다. 이는 실제 코딩 영역에서 프레임에 따라 주기성이 달라지는 현상을 정량화한다. 비동질 모델에서도 R(k,n) 비율을 정의해 프레임별 주기성 변화를 추적한다.
실험에서는 세 가지 데이터셋을 사용한다. (1) 길이 1000의 완전 무작위 서열은 주기성이 없으며, 계산된 R(n) 값이 빠르게 0에 수렴한다. (2) 3‑염기 주기를 인위적으로 삽입한 합성 서열에서는 A 염기에 대한 p_A(n,3) 가 다른 염기보다 현저히 높고, R(n) 값이 지속적으로 1에 근접한다. (3) 실제 인간 디스토핀 mRNA(코딩)와 BNGF 유전자(비코딩) 서열을 적용했을 때, 코딩 서열에서는 특정 프레임에서 높은 R(k,n) 값을 보이며, 비코딩 서열에서는 전반적으로 낮은 값을 보여 모델이 코딩/비코딩 영역을 구분함을 확인했다.
모델 파라미터는 경험적 전이 횟수 N(i→j)와 체류 횟수 N(i→j,m)를 이용해 식 (29) 로 추정한다. 초기조건은 첫 10 사이클(길이 3) 데이터를 사용해 추정하고, 이후 각 사이클마다 핵심 행렬 C(k,m)을 갱신한다.
결론적으로, 이 논문은 DNA 3‑염기 주기를 반마코프 체인으로 정량화하고, 주기성 강도를 나타내는 R(n)·R(k,n) 지표를 제안함으로써 기존 Fourier 변환이나 HMM 기반 방법보다 체류 시간까지 고려한 더 정교한 주기성 분석을 가능하게 한다. 또한 비동질 모델을 통해 코딩 프레임별 특성을 별도로 분석할 수 있어, 엑손‑인트론 경계 탐지 및 유전자 구조 예측에 실용적인 도구가 될 전망이다.
**
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기