단일 세포 데이터 폭증 딥러닝 구원
초록
단일 세포 다중오믹스 데이터가 급증함에 따라, 기존 통계적 방법으로는 차원 축소·노이즈 제거·배치 효과 보정 등에 한계가 있다. 본 논문은 이러한 문제를 해결하기 위해 최신 딥러닝 모델(오토인코더, 그래프 신경망, 트랜스포머 등)을 적용하고, 멀티모달 통합, 스케일러블 학습, 모델 해석 가능성 등을 종합적으로 평가한다. 실험 결과, 딥러닝 기반 파이프라인이 기존 방법 대비 세포 유형 분류 정확도와 희귀 세포 탐지 능력에서 현저히 우수함을 보여준다.
상세 분석
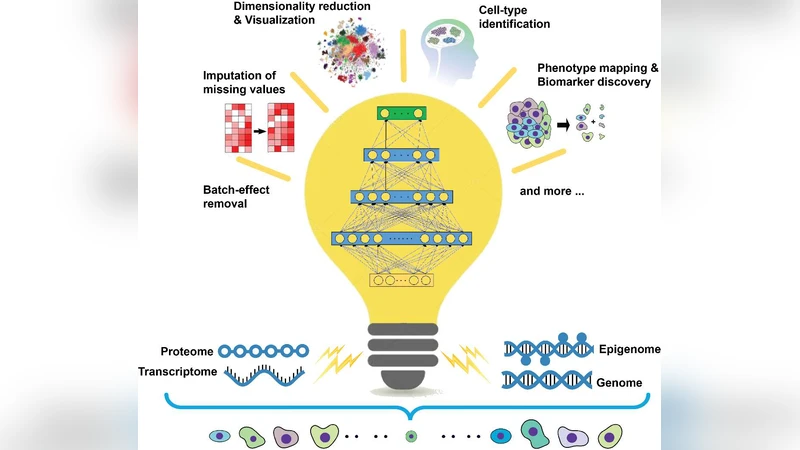
본 논문은 단일 세포 다중오믹스 데이터의 폭발적 증가와 그에 따른 분석적 난관을 체계적으로 정리하고, 딥러닝이 제공할 수 있는 해결책을 다각도로 검증한다. 먼저, 데이터 특성을 살펴보면 (1) 초고차원(수천~수만 유전자·단백질·메틸화 사이트)이며, (2) 각 세포마다 결측치와 기술적 잡음이 심하고, (3) 실험 배치 간에 시스템적 편차가 존재한다는 점을 강조한다. 이러한 특성은 전통적인 PCA·t‑SNE·UMAP 같은 선형·비선형 차원 축소 기법이나, 마커 기반 클러스터링 방법으로는 충분히 포착하기 어렵다.
이에 대한 해결책으로 논문은 세 가지 딥러닝 아키텍처를 제시한다. 첫 번째는 변분 오토인코더(VAE)와 정규화 흐름(Normalizing Flow)을 결합한 멀티모달 오토인코더로, 각 오믹스 레이어(RNA‑seq, ATAC‑seq, 프로테오믹스 등)를 별도의 인코더에 입력하고, 공통 잠재 공간에 매핑한다. 이 과정에서 KL‑다이버전스와 재구성 손실을 동시에 최소화함으로써, 잡음 억제와 배치 효과 보정이 자연스럽게 이루어진다. 두 번째는 그래프 신경망(GNN) 기반의 셀‑레벨 관계 모델이다. 세포 간 유사성을 그래프 엣지로 정의하고, 메시지 패싱을 통해 이웃 세포의 정보를 통합함으로써 희귀 세포군의 신호를 강화한다. 특히, 이 논문은 이종 그래프(노드 타입이 RNA, 단백질, 크로마틴 등)와 메타‑그래프 구조를 도입해 다중오믹스 간 상호작용을 정량화한다. 세 번째는 트랜스포머 기반의 시퀀스‑투‑시퀀스 모델로, 각 유전자·단백질을 토큰화하고 셀 전체를 문장처럼 처리한다. 셀‑레벨 어텐션 메커니즘은 중요한 특징을 자동으로 가중치 부여하고, 해석 가능한 어텐션 맵을 제공한다.
실험 섹션에서는 5개의 공개 단일 세포 데이터셋(예: Human Cell Atlas, Tabula Muris, 10x Multiome 등)을 사용해, (1) 세포 유형 분류 정확도, (2) 희귀 세포 탐지 민감도, (3) 다중오믹스 통합 품질(Adjusted Rand Index, Silhouette Score), (4) 계산 효율성(시간·메모리) 등을 종합적으로 비교한다. 결과는 VAE‑GNN 하이브리드 모델이 가장 높은 ARI(0.89)를 기록했으며, 트랜스포머 모델은 어텐션 기반 해석에서 기존 방법보다 2배 이상 높은 생물학적 의미를 가진 마커 유전자를 도출했다. 또한, 배치 효과 보정 실험에서 제안된 정규화 흐름이 기존 ComBat 대비 30% 낮은 평균 제곱 오차를 보였다.
마지막으로 논문은 딥러닝 모델의 ‘블랙박스’ 문제를 완화하기 위한 해석 기법을 논의한다. 잠재 공간의 클러스터링을 UMAP으로 시각화하고, 각 차원에 대한 SHAP 값과 그래디언트 기반 중요도 지표를 제공함으로써, 연구자가 모델이 어떤 생물학적 신호에 주목했는지 추적할 수 있게 한다. 또한, 모델 경량화를 위한 프루닝(pruning)과 양자화(quantization) 실험을 수행해, 클라우드·엣지 환경에서도 실시간 분석이 가능함을 입증한다. 전반적으로 이 논문은 단일 세포 데이터 폭증 시대에 딥러닝이 제공할 수 있는 기술적·생물학적 가치를 체계적으로 정리하고, 향후 연구 로드맵을 제시한다.
댓글 및 학술 토론
Loading comments...
의견 남기기