프로토폴드 II: 효율적인 케네토스태틱 단백질 접힘 모델과 고속 구현
초록
Protofold II는 기존 KCM 기반 단백질 접힘 시뮬레이션에 용매 효과와 엔트로피를 반영한 새로운 자유에너지 모델을 도입하고, 해시 테이블·트리 구조 등 선형 복잡도의 알고리즘을 설계해 CPU와 GPU에서 최대 100배 가속을 달성한다. 이를 통해 수천 개 원자를 갖는 대형 단백질도 실시간에 가까운 속도로 2차·3차 구조 형성을 관찰할 수 있다.
상세 분석
Protofold II는 두 가지 차원에서 기존 Protofold I을 크게 개선한다. 첫째, 물에 용해되는 단백질의 핵심 구동력인 소수성 효과를 정량화하기 위해 선형 암시적 용매 모델(ASA 기반)을 채택하고, 이를 위한 오프셋 표면 열거 기법을 제안한다. 이 방법은 정확한 SASA 계산(구면 교차 면적)보다 10~100배 빠르면서도, 기존 확률적 근사(LCPO, 63번 논문)보다 오차를 현저히 줄인다. 두 번째 개선은 알고리즘적 복잡도이다. 기존 O(n²) 구조의 원자 간 상호작용 계산을 3D 해시 그리드와 트리 기반 거리 탐색으로 O(n)으로 낮추었다. 특히, 절단 거리(cut‑off)를 이용해 무시 가능한 상호작용을 사전에 필터링하고, 토크 전파에 prefix‑sum을 활용해 관절 토크 계산을 선형 시간에 수행한다.
병렬화 측면에서는 OpenMP 기반 CPU 구현이 10배 수준의 가속을 보였으며, GPU 구현에서는 데이터‑병렬 SASA 열거와 힘 계산을 SIMT 아키텍처에 맞게 메모리 계층을 최적화함으로써 최대 100배 가속을 달성했다. 이때, 원자 좌표와 해시 인덱스를 연속 메모리 블록에 배치하고, 원자‑원자 쌍 탐색을 warp‑level 협업으로 수행해 메모리 대역폭을 효율적으로 활용했다.
물리 모델 측면에서는 단백질을 개방형 링크 체인으로 모델링하고, 각 회전 관절(φ, ψ, χ)을 토크에 비례하는 각도 변화로 업데이트한다. 전통적인 MD가 1 fs 이하의 타임스텝을 요구하는 반면, KCM은 1 ps 수준의 가상 타임스텝을 사용해 수천 배 빠른 수렴을 보인다. 전자기력(쿨롱)과 반데르발스 힘은 기존과 동일하게 처리하면서, 새롭게 도입된 용매 자유에너지와 그 기울기(힘)를 동시에 고려해 전체 자유에너지의 최소화를 목표로 한다.
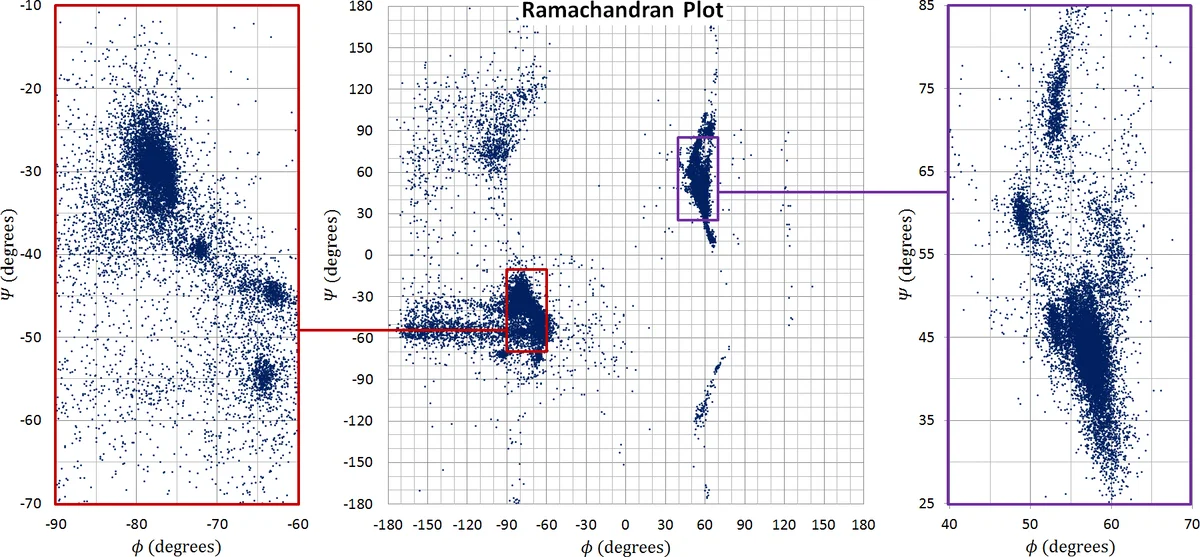
실험 결과는 2차 구조(α‑헬릭스, β‑시트)의 형성 시점과 3차 구조(핵심 소수성 코어)의 완성 시점을 정량적으로 보여준다. 특히, 용매 효과를 포함했을 때 물에 친화적인 표면이 먼저 형성되고, 비극성 잔기가 내부로 몰입하는 전형적인 소수성 주도 메커니즘이 재현된다. 대형 단백질(>3000 원자)에서도 시뮬레이션이 수시간 내에 완료되어, 기존 MD 기반 방법이 수일~수주가 걸리던 문제를 크게 완화한다.
요약하면, Protofold II는 물리‑화학적 정확성을 크게 손상시키지 않으면서, 선형 시간 복잡도와 GPU 가속을 통해 실용적인 대규모 단백질 접힘 시뮬레이션을 가능하게 만든 혁신적인 프레임워크라 할 수 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기