유전자 집합 배깅: 고처리 데이터에서 생물학적 해석의 재현성을 평가하는 새로운 방법
본 연구는 기존 유전자 집합 분석의 불안정성을 해결하기 위해 “유전자 집합 배깅(gene set bagging)”이라는 부트스트랩 기반 절차를 제안한다. 원본 데이터와 동일한 크기의 샘플을 복원추출하고, 각 부트스트랩 샘플에 대해 차등 발현·메틸화 분석 및 유전자 집합 풍부도 검정을 수행한다. 반복된 검정 결과로 각 유전자 집합이 새로운 실험에서 유의미하게 나타날 확률, 즉 복제 확률 R을 추정한다. 흡연 관련 전사체 데이터와 뇌 조직 메틸화 …
저자: Andrew E. Jaffe, John D. Storey, Hongkai Ji
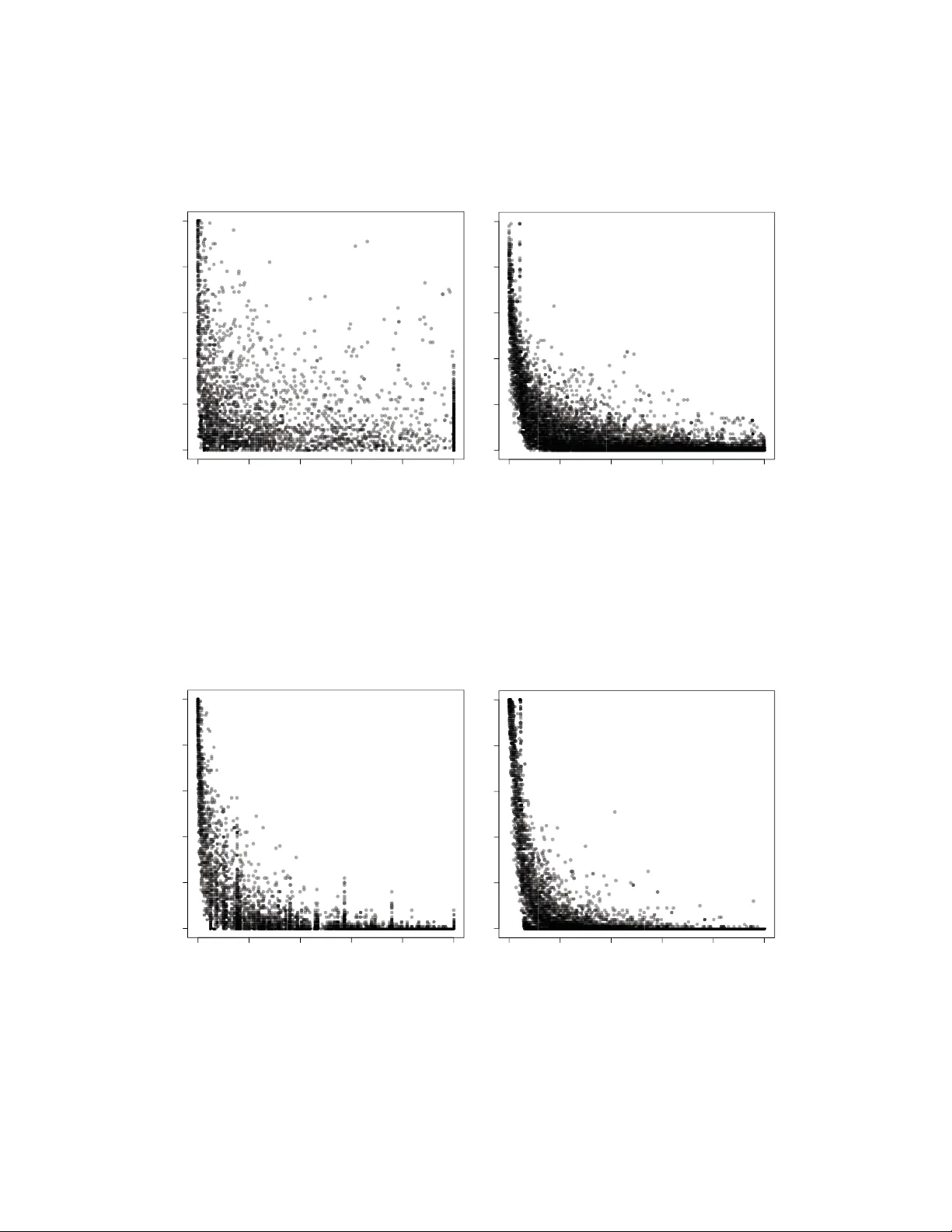
본 논문은 고처리 유전체 데이터에서 흔히 사용되는 유전자 집합 분석(gene set enrichment analysis, GSEA) 및 하이퍼지오메트리 검정이 실제 재현성 측면에서 얼마나 불안정한지를 지적하고, 이를 보완하기 위한 새로운 통계적 절차인 “유전자 집합 배깅(gene set bagging)”을 제안한다. 저자들은 먼저 기존 분석 흐름—각 유전자에 대한 차등 발현·메틸화 검정 → 유전자 집합 풍부도 검정—이 두 단계 모두에서 변동성을 내포하고 있음을 설명한다. 특히, 유전자 순위 자체가 데이터 샘플링에 민감하게 변하고, 이러한 순위 변동이 곧바로 유전자 집합 수준의 p‑값에 영향을 미친다. 따라서 단일 데이터셋에 대한 p‑값만으로는 해당 집합이 다른 연구에서도 동일하게 유의미할지를 판단하기 어렵다.
이를 해결하기 위해 저자들은 전체 분석 파이프라인을 블랙박스로 보고, 원본 데이터에서 복원추출(bootstrapping)된 B = 100개의 샘플을 생성한다. 각 부트스트랩 샘플에 대해 동일한 차등 발현 검정(예: empirical Bayes t‑statistic)과 유전자 집합 풍부도 검정(예: Wilcoxon rank‑sum, hypergeometric test)을 수행한다. 각 반복에서 얻어진 유전자 집합 p‑값(p*_{b,l})이 사전 정의된 유의수준 α(보통 0.05) 이하인지 여부를 기록하고, 이를 통해 복제 확률 R_l = (1/B)∑_{b=1}^B I(p*_{b,l}<α) 를 추정한다. R_l은 “이 유전자 집합이 새로운 독립 실험에서도 유의미하게 검출될 확률”을 의미한다.
논문은 두 가지 실제 데이터에 이 방법을 적용한다. 첫 번째는 흡연 여부에 따른 구강 상피 전사체 데이터(GSE17913)이며, 40명의 흡연자와 39명의 비흡연자를 비교한다. 데이터 전처리 단계에서는 RMA 정규화와 surrogate variable analysis(SVA)를 통해 배치 효과를 보정한다. 차등 발현 검정 후 q‑값<0.05인 유전자를 선정하고, GO 기반 전사체 풍부도 검정을 수행한다. 이후 gene set bagging을 적용한 결과, 전통적인 p‑값 기준으로는 비유의적이지만 R > 0.8인 8개의 GO 카테고리가 발견되었다. 이들 카테고리는 흡연과 관련된 인산화 과정 및 대사 조절에 관여하는 것으로, 기존 분석에서는 놓칠 수 있었던 생물학적 신호를 제공한다.
두 번째는 뇌 조직 간 DNA 메틸화 차이를 분석한 데이터(GSE15745)이다. 전두엽과 측두엽을 각각 131, 126개의 샘플로 비교하고, plate, tissue bank, 성별, 연령을 공변량으로 포함한 선형 모델을 적용한다. 메틸화 차이가 있는 CpG를 q‑값<0.05로 선정한 뒤, 해당 유전자를 GO에 매핑하고 gene set bagging을 수행한다. 결과적으로 p < 0.05이지만 R = 0인 소수의 작은 유전자 집합이 존재했으며, 이는 통계적으로는 유의하지만 실제 재현성이 전혀 없음을 의미한다. 반대로 p > 0.05이지만 R > 0.8인 집합도 다수 확인되어, 전통적인 p‑값 기준만으로는 놓칠 수 있는 의미 있는 생물학적 경로를 드러냈다.
시뮬레이션 연구에서는 두 가지 시나리오를 설정했다. 첫 번째는 100개의 유전자를 대상으로 신호를 삽입하고, 1,000개의 독립 데이터셋을 생성해 각 데이터셋에 대해 GSEA와 gene set bagging을 수행했다. 두 번째는 500개의 유전자를 대상으로 신호를 삽입하고, 100쌍의 데이터셋을 비교했다. 두 경우 모두 복제 확률 R_l이 실제 반복 실험에서 해당 집합이 유의미하게 검출된 비율과 높은 상관관계를 보였다. 이는 R_l이 “미래 실험에서 재현될 확률”을 정량적으로 추정하는 데 유효함을 입증한다.
논문은 R과 전통적인 p‑값 사이의 관계를 표 1과 그림 1‑3을 통해 시각화한다. R = 1이면서 p > 0.05인 경우는 “매우 안정적이지만 비유의적”으로 해석되며, 이는 연구자가 관심 있는 생물학적 현상이 통계적으로는 검출되지 않았지만 재현성이 높아 추가 검증이 가치 있음을 시사한다. 반대로 R = 0이면서 p < 0.05인 경우는 “불안정하지만 통계적으로 유의”로, 이러한 집합은 실제로는 실험 재현성이 낮아 과대해석을 경계해야 한다.
결론적으로, 유전자 집합 배깅은 기존의 단일 p‑값 기반 GSEA에 비해 두 가지 중요한 장점을 제공한다. 첫째, 데이터 내 변동성을 직접 반영한 복제 확률 R을 제공함으로써 결과의 신뢰성을 정량화한다. 둘째, R과 p‑값을 동시에 고려함으로써 “통계적 유의성”과 “생물학적 재현성” 사이의 균형을 잡을 수 있다. 저자들은 이 방법이 다양한 고처리 플랫폼(RNA‑seq, ATAC‑seq, 메틸화 배열 등)에 적용 가능하며, 특히 샘플 수가 제한적이거나 잡음이 큰 경우에 유용할 것이라고 제언한다. 향후 연구에서는 부트스트랩 반복 수 최적화, 다중 그룹 설계 확장, 그리고 베이지안 프레임워크와의 통합 등을 통해 방법론을 더욱 정교화할 여지가 있다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기