MOF 연구를 위한 컴퓨팅 도구와 예측 전략
초록
이 리뷰는 금속‑유기 골격체(MOF)의 구조·물성 예측에 활용되는 양자화학 및 고전분자 시뮬레이션 방법을 정리한다. 결정구조 예측, 가상 MOF 데이터베이스 구축, 기공·표면적 등 기하학적 특성 계산, 기계·열·광학·전기적 물성 평가, 그리고 가스 흡착·분리 시뮬레이션까지 전 과정을 다루며, 현재의 한계와 향후 연구 방향을 제시한다.
상세 분석
본 논문은 MOF 분야에서 컴퓨팅이 차지하는 역할을 체계적으로 정리한다. 먼저 결정구조 예측 방법을 살펴보면, 전통적인 시뮬레이티드 어닐링, 유전 알고리즘, 그리고 AASBU(Automated Assembly of Secondary Building Units)와 같은 SBUs 기반 접근법을 소개한다. 특히, AASBU는 “스티키 원자” 상호작용을 이용해 SBUs가 자가조립하도록 하여 실제 실험 구조를 재현하거나 새로운 구조를 제안한다. 그러나 계산 비용이 높아 대규모 스크리닝에는 부적합하다. 이를 보완하기 위해 토폴로지 장식 전략과 이소레터리컬(iso‑reticular) 접근법이 제시된다. 전자는 수학적 네트를 SBUs에 매핑해 가능한 토폴로지를 전부 탐색하고, 후자는 기존 MOF의 토폴로지를 유지하면서 리간드 길이와 기능을 변형해 새로운 구조를 설계한다. 두 방법 모두 DFT 혹은 고전 포스필드로 에너지 최소화를 수행해 상대 안정성을 평가한다.
대규모 가상 MOF 데이터베이스 구축에서는 Wilmer 그룹이 제시한 “LEGO‑like” 빌딩 블록 조합 방식이 핵심이다. 실험에 보고된 금속 클러스터와 유기 리간드를 사전 정의하고, 기하학적 결합 규칙에 따라 조합함으로써 수십만 개의 가상 구조를 자동 생성한다. 이러한 데이터베이스는 고속 전산 스크리닝을 가능하게 하며, 물리·화학적 특성(표면적, 기공 부피, 밀도 등)을 사전 계산해 후보 물질을 선별한다.
기하학적 특성 평가에서는 Connolly 표면, Voronoi 분할, 그리고 고급 지표(예: 프랙탈 차원, 접근성 지수) 등이 논의된다. 특히, 기공 크기 분포와 표면적은 가스 흡착 용량을 예측하는 데 핵심 변수이며, 이를 위한 Zeo++와 같은 오픈소스 툴이 널리 사용된다.
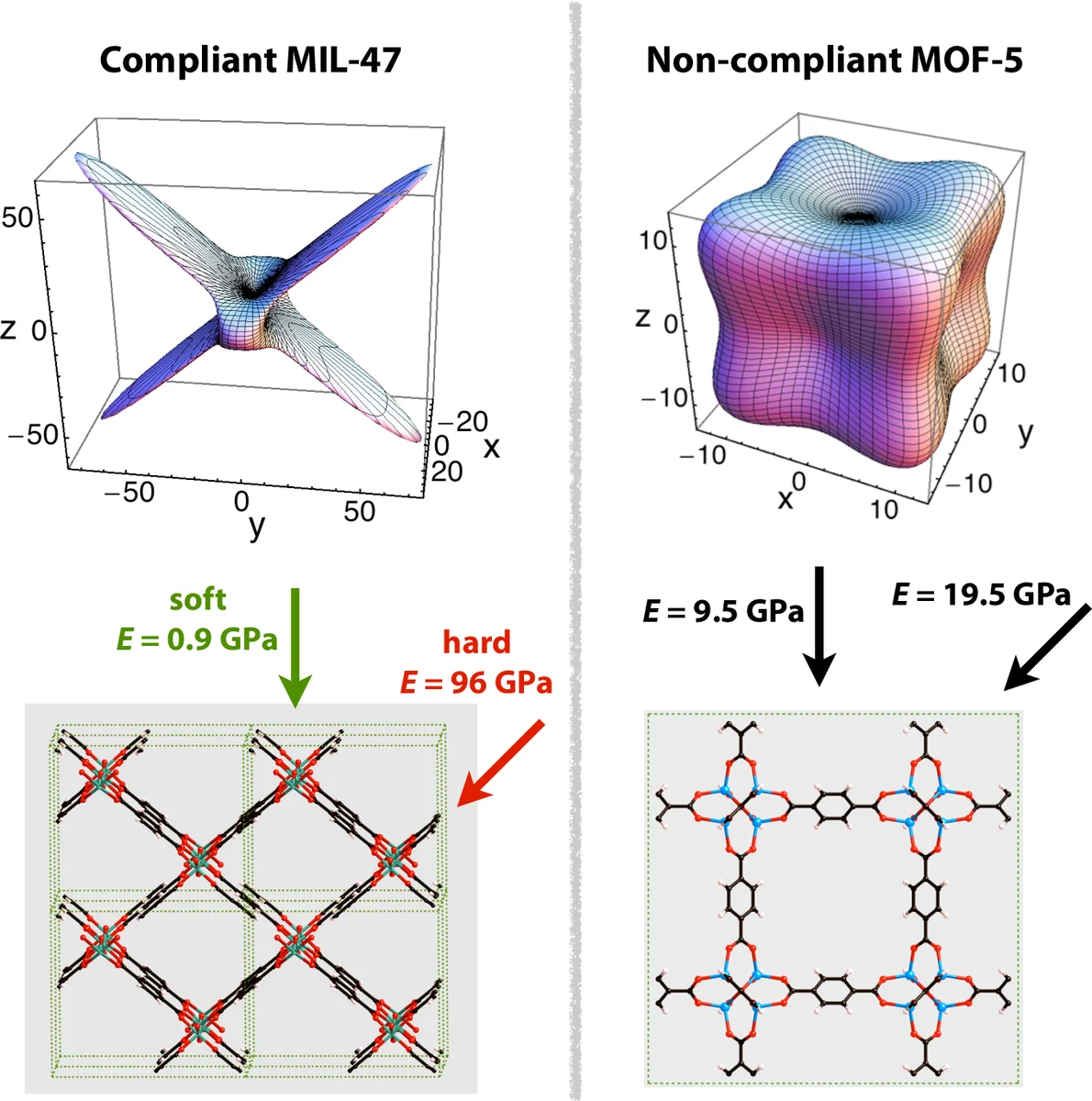
물성 측면에서는 고전 MD와 DFT 기반 탄성 상수 계산을 통해 MOF의 압축성, 전단강도, 열팽창 계수를 얻는다. 열전도와 열용량은 포논 계산이나 분자 동역학 시뮬레이션으로 추정되며, 전자·광학 특성은 밴드 구조와 TD‑DFT를 통해 분석한다.
흡착 시뮬레이션은 Grand Canonical Monte Carlo(GCMC) 방법이 주류이며, 고전 포스필드와 오픈 메탈 사이트를 위한 특수 파라미터가 필요하다. 유연 MOF의 경우, GCMC와 MD를 결합한 “Hybrid” 접근법이 제안되며, 이는 구조 변형이 흡착 등온선에 미치는 영향을 포착한다. 또한, 다중 성분 흡착·분리, 화학흡착(chemisorption) 모델링, 실험 데이터와의 비교 방법론도 상세히 다룬다.
마지막으로, 결함·무질서 모델링, 고속 스크리닝을 위한 데이터베이스 구축, 그리고 자극 반응형(MOF‑responsive) 설계 등 미래 과제가 제시된다. 전반적으로, 양자·고전 시뮬레이션이 실험과 긴밀히 연계될 때 MOF 설계 효율이 크게 향상될 수 있음을 강조한다.
댓글 및 학술 토론
Loading comments...
의견 남기기