실리케이트 양극재의 사면체 구조 탐색: 모티프 네트워크 접근법
본 연구는 모티프‑네트워크 탐색 기법을 이용해 A₂MSiO₄(Li, Na와 Mn, Fe, Co)의 사면체 구조를 전산적으로 조사하였다. 기존 보고된 구조를 모두 재현함과 동시에 에너지적으로 거의 동등한 새로운 1D·2D·3D M‑Si‑O 골격을 가진 다수의 구조를 발견하였다. 특히 Na 기반 시스템에 대해 최초로 최저에너지 구조를 제시하고, 구조 선호도와
초록
본 연구는 모티프‑네트워크 탐색 기법을 이용해 A₂MSiO₄(Li, Na와 Mn, Fe, Co)의 사면체 구조를 전산적으로 조사하였다. 기존 보고된 구조를 모두 재현함과 동시에 에너지적으로 거의 동등한 새로운 1D·2D·3D M‑Si‑O 골격을 가진 다수의 구조를 발견하였다. 특히 Na 기반 시스템에 대해 최초로 최저에너지 구조를 제시하고, 구조 선호도와 안정성에 영향을 주는 화학적·결정학적 지표들을 제안하였다.
상세 요약
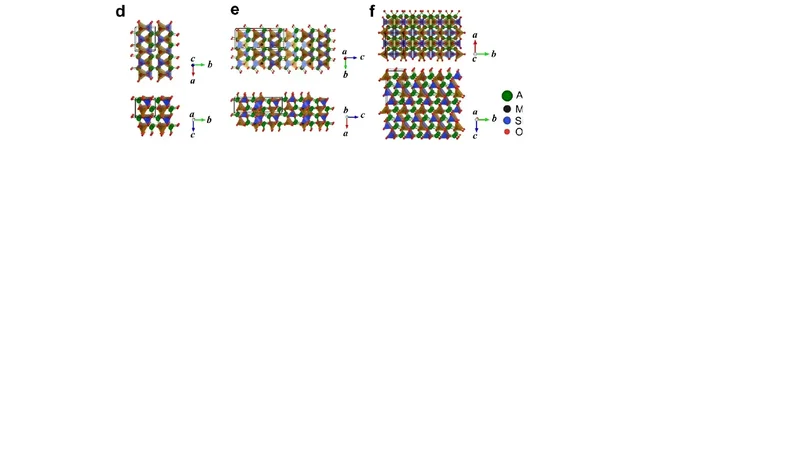
본 논문은 전이금속 옥시실리케이트 A₂MSiO₄(Li/Na와 M = Mn, Fe, Co)의 배터리 양극재로서의 구조적 다양성을 체계적으로 탐색하기 위해 ‘모티프‑네트워크’라는 새로운 탐색 프레임워크를 도입하였다. 이 방법은 사면체 연결망을 기본 모티프로 삼아, 가능한 모든 사면체 토폴로지를 자동 생성하고, 첫 원리 계산(Density Functional Theory, DFT)으로 에너지를 평가한다. 기존에 알려진 구조(예: α‑, β‑, γ‑형)뿐 아니라, 에너지 차이가 수 meV 수준으로 거의 동일한 수십 종의 새로운 구조를 발견했으며, 이들을 M‑Si‑O 골격이 1차원(선형), 2차원(층상) 혹은 3차원(입체)으로 연결되는 형태로 분류하였다.
에너지 분포 분석 결과, 동일 화학식 내에서도 다중의 거의 동등한 구조가 존재함을 확인했으며, 이는 실제 합성 과정에서 온도·압력·합성 전구체 등에 따라 다양한 상이 공존할 가능성을 시사한다. 구조 선호도에 영향을 주는 주요 지표로는 (1) A‑이온(Li/Na)의 크기와 전하밀도, (2) 전이금속 M의 전자구조(특히 d‑오비탈 점유), (3) Si‑O 사면체의 왜곡 정도를 정량화한 ‘tetrahedral distortion index’를 제시하였다. 특히 Na⁺는 Li⁺보다 반경이 크고 전하밀도가 낮아, 사면체 네트워크 내에서 더 큰 공간적 여유를 제공한다. 이로 인해 Na‑계열에서는 2D·3D 골격이 동시에 안정화되는 경향이 뚜렷하게 나타났으며, Li‑계열에서는 1D·2D 구조가 주로 우세했다.
Na 기반 시스템에 대해서는 기존 문헌이 거의 없었으나, 본 연구는 Na₂MSiO₄(M = Mn, Fe, Co)의 최저에너지 구조를 각각 예측하고, 그 구조가 기존 Li‑계열과는 다른 독특한 3D 골격을 형성함을 보고하였다. 이는 Na‑이온이 큰 구조적 자유도를 제공함에 따라 새로운 전이금속‑실리콘‑산소 네트워크가 형성될 수 있음을 의미한다. 또한, 에너지 차이가 작아 실험적으로는 다중 상이 공존할 가능성이 높으며, 이는 배터리 성능(전압, 용량, 사이클 안정성) 최적화에 활용될 수 있는 설계 변수로 작용한다.
전반적으로, 모티프‑네트워크 탐색은 전이금속 옥시실리케이트와 같은 복합 산화물의 구조 공간을 효율적으로 샘플링할 수 있는 강력한 도구임을 입증하였다. 향후 이 방법을 다른 다원소 시스템(예: 폴리머·무기 복합체)에도 확장한다면, 고성능 에너지 저장·전환 소재의 설계에 큰 기여를 할 것으로 기대된다.
📜 논문 원문 (영문)
🚀 1TB 저장소에서 고화질 레이아웃을 불러오는 중입니다...