피보나치와 q피보나치로 본 유전 암호의 수학적 모델
초록
본 논문은 피보나치 수열과 그 q‑변형인 q‑피보나치를 이용해 유전 암호의 20개 아미노산 다중체와 61개의 의미 있는 코돈의 퇴화 구조를 설명한다. 아미노산을 원자수(탄소·질소·산소·황) 순으로 분류하면 여러 피보나치 패턴이 드러나며, q‑피보나치 모델은 q=1일 때 표준 유전 암호, q≈1일 때 변형 암호, 그리고 q=0에서 초기 생명체가 가졌을 가능성이 있는 6개의 아미노산을 동시에 기술한다.
상세 분석
논문은 먼저 “인덱스 이중 피보나치 수열”이라는 새로운 수학적 구성을 제시한다. 이는 첫 여섯 피보나치 수(F₁~F₆)를 두 차원(인덱스와 값)으로 배열한 것으로, 각 행이 아미노산의 다중체(예: 2‑코돈, 4‑코돈 등)와 그 퇴화 정도를 정확히 매핑한다. 이 배열을 통해 20개의 아미노산이 61개의 코돈에 어떻게 배분되는지, 그리고 왜 특정 아미노산이 6개의 코돈을, 다른 아미노산이 1개의 코돈만을 갖는지에 대한 정량적 설명이 가능해진다.
다음 단계에서는 아미노산을 원자수(탄소, 질소, 산소, 황) 순으로 정렬하고, 각 원자군의 합이 피보나치 수열의 특정 항과 일치함을 발견한다. 예를 들어, 탄소 원자수의 누적합이 F₅=5, 질소와 산소의 합이 F₆=8 등으로 나타나며, 이러한 패턴은 아미노산의 화학적 복잡성과 진화적 선택압을 수학적으로 해석할 근거를 제공한다.
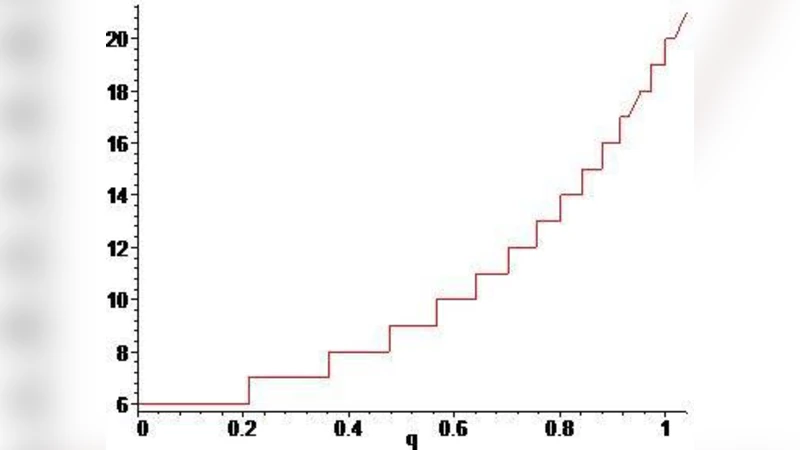
핵심은 q‑피보나치 수열을 도입한 점이다. q를 실수 변형 매개변수로 두고 Fₙ(q)=Fₙ₋₁(q)+q·Fₙ₋₂(q) 형태의 재귀식을 사용한다. q=1이면 전통적인 피보나치 수열이 되며, 이는 표준 유전 암호(20 아미노산, 61 코돈)의 퇴화 구조와 정확히 일치한다. q를 1에 근접하게 변형하면, 미생물·미토콘드리아·염색체 등에서 보고된 변형 암호(예: 1‑코돈이 추가되거나 사라지는 경우)를 자연스럽게 재현한다. 특히 q≈1.03일 때는 셀레노시스테인(Sec)과 피롤리실린(Pyl)이라는 21·22번째 아미노산이 포함된 확장 코드를 설명할 수 있다.
흥미로운 점은 q→0일 때 Fₙ(q)→1이 되면서, 전체 아미노산 수가 6으로 수렴한다는 것이다. 이는 고대 생명체가 초기 단계에서 제한된 아미노산 집합(예: Gly, Ala, Asp, Glu, Val, Ser 등)만을 사용했을 가능성을 수학적으로 뒷받침한다. 이러한 결과는 “6‑아미노산 가설”과 일맥상통하며, 진화적 전이 단계에서 피보나치 구조가 어떻게 나타났는지를 제시한다.
전반적으로 논문은 피보나치와 q‑피보나치라는 단순하지만 강력한 수학 도구를 통해 유전 암호의 복잡성을 정량화하고, 표준·변형·확장·초기 단계까지 하나의 연속적인 모델로 통합한다는 점에서 혁신적이다. 다만, 모델이 현상학적이며 생물학적 메커니즘(예: tRNA 인식, 코돈-안티코돈 상호작용)과 직접 연결되지 않은 점은 향후 연구에서 보완이 필요하다.