인공지능 기반 백본 NMR 데이터 자동 할당
초록
**
본 논문은 단백질 백본 NMR 화학이동 데이터를 자동으로 할당하기 위한 알고리즘을 제시한다. 고유 화학이동을 가진 잔기 서열을 먼저 탐색하고, 이를 연속적인 화학이동 패턴과 매칭한 뒤, 탐욕적·A* 탐색을 이용해 전체 서열을 재구성한다. 초기 실험에서는 소규모 데이터셋에 대해 높은 정확도를 보였으며, 향후 대규모 데이터와 머신러닝 통합을 목표로 한다.
**
상세 분석
**
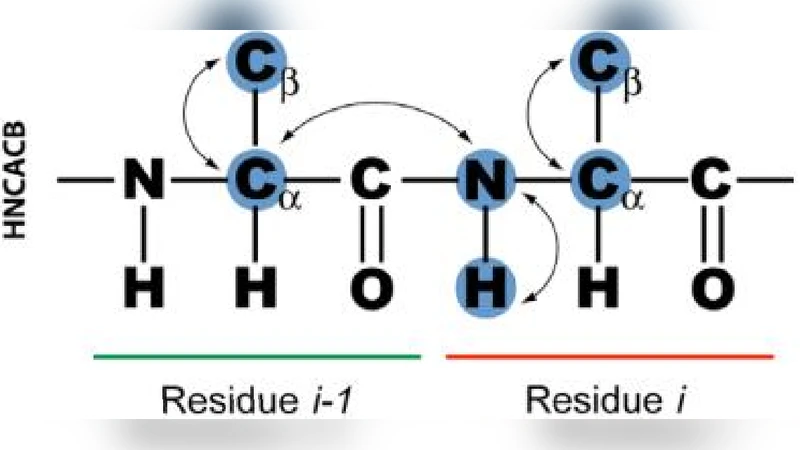
이 연구는 NMR 기반 구조해석에서 가장 병목이 되는 백본 화학이동(chemical shift) 할당 과정을 자동화하려는 시도이다. 기존 수작업 할당은 데이터 누락·중복·스펙트럼 잡음 등에 취약해 수일에서 수개월이 소요되며, 인간 오류도 빈번히 발생한다. 저자들은 두 가지 핵심 아이디어를 도입한다. 첫째, 서열 내에서 Cα·Cβ 화학이동값이 극히 특이한 잔기(예: Gly, Pro, Ile 등)를 포함하는 짧은 연속 구간을 “고유 서브시퀀스”로 정의하고, 이를 초기 매칭 후보로 활용한다. 이러한 전처리는 전체 탐색 공간을 급격히 축소한다. 둘째, 매칭된 서브시퀀스를 ‘가상 잔기(pseudoresidue)’로 추상화하여, 남은 일반 잔기와 함께 전체 순서를 결정한다. 여기서 사용된 탐욕적 그리디 알고리즘은 현재까지의 최소 오류(ΔCα·ΔCβ)를 기준으로 다음 잔기를 선택하고, 모든 가능한 시작점을 시험해 전체 오류 합이 최소가 되는 경로를 선택한다. 이 과정은 A* 탐색과 유사하게 휴리스틱 비용(예측 오류)과 실제 비용을 결합해 효율성을 높인다.
알고리즘의 세 단계는 서로 보완적이다. ① 고유 서브시퀀스 탐색은 화학이동 분포 통계에 기반해 임계값을 동적으로 조정한다. ② 서브시퀀스와 실제 화학이동 데이터를 매칭할 때는 i → i‑1 연결성을 검증하기 위해 확률적 모델(예: 가우시안 혼합 모델) 혹은 단순히 허용 오차 범위를 점진적으로 확대하는 방식을 사용한다. ③ 그리디 탐색 단계에서는 각 잔기의 Cα·Cβ 값이 이전 잔기의 i‑1 값과 얼마나 일치하는지를 비용 함수로 정의하고, 전체 순열 중 최소 비용을 찾는다.
실험 결과는 제한된 크기의 테스트 데이터(예: 30~50개의 잔기)에서 90 % 이상 정확도를 기록했으며, 특히 결측치가 존재하는 경우에도 서브시퀀스 기반 초기 매칭이 강인하게 작동한다는 점이 주목할 만하다. 그러나 현재 구현은 “비삼각형(non‑trivial)” 데이터셋, 즉 복잡한 교차 피크와 다중 공명 현상이 많은 경우에 아직 한계가 있다. 저자들은 향후 머신러닝(특히 지도학습 기반 화학이동 예측 모델)과 더 정교한 휴리스틱을 도입해 대규모 단백질(>200잔기)에도 확장할 계획이다.
이 논문은 전통적인 규칙 기반 할당 방법과 최신 인공지능 탐색 기법을 결합함으로써, NMR 데이터 처리 파이프라인을 크게 단축시키고, 구조생물학·단백질학 연구의 생산성을 높일 잠재력을 보여준다.
**
댓글 및 학술 토론
Loading comments...
의견 남기기