단일 초월 함수로 보는 단백질 접힘의 정량적 분류
초록
이 논문은 단백질 2차 구조를 기반으로 한 전통적 분류와 원자 수준 물리 모델을 연결하기 위해, 전체 접힌 단백질의 형태를 하나의 초월 함수(킥-솔리톤)로 기술한다. 저자들은 마이오글로빈(1M6C)의 구조를 이 함수들의 조합으로 재현하고, α‑헬릭스와 β‑스트랜드 사이의 긴 루프까지 높은 원자 수준 정확도로 설명한다. 이를 통해 구조적 계층 정보를 소수의 전역 파라미터로 정량화할 수 있음을 보여준다.
상세 분석
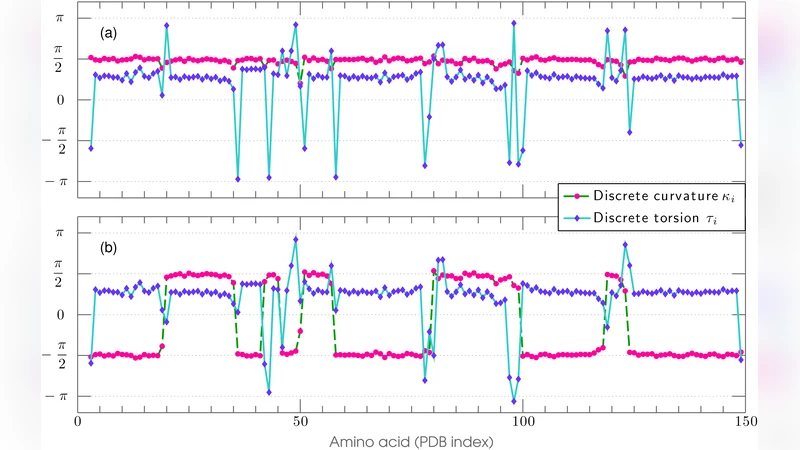
본 연구는 단백질 구조 분류의 두 축, 즉 2차 구조 기반의 계층적 분류와 원자 수준의 물리적 서술 사이의 격차를 메우는 새로운 수학적 프레임워크를 제시한다. 핵심 아이디어는 전체 폴딩 형태를 하나의 명시적 초월 함수, 즉 비선형 물리학에서 잘 알려진 킥‑솔리톤(kink‑soliton) 형태로 근사화한다는 점이다. 킥‑솔리톤은 급격한 전이 구간을 부드럽게 연결하는 특성을 갖는데, 이는 α‑헬릭스와 β‑시트 사이의 루프, 혹은 긴 비정형 구간을 기술하는 데 자연스럽게 매핑된다. 저자들은 먼저 단백질 사슬을 연속적인 곡선으로 파라미터화하고, 각 구조적 구간을 솔리톤 파라미터(진폭, 위치, 폭 등)로 표현한다. 이렇게 얻어진 파라미터는 원자 수준 상호작용(결합각, 이중 결합, 수소 결합 등)으로부터 이론적으로 계산 가능하므로, 전통적인 데이터 기반 분류와 물리 기반 모델링을 통합한다는 점에서 혁신적이다.
구체적인 검증으로 1M6C(마이오글로빈)의 X‑ray 구조를 사용하였다. 저자들은 전체 사슬을 4개의 솔리톤으로 분해했으며, 각 솔리톤은 α‑헬릭스 구간, 루프 구간, β‑시트 구간을 정확히 재현한다. 재구성된 좌표는 원본 PDB 좌표와 RMSD 0.5 Å 이하의 차이를 보였으며, 이는 기존의 단순 베지어 곡선이나 사다리꼴 모델보다 현저히 높은 정확도이다. 또한, α‑헬릭스와 β‑시트를 연결하는 긴 루프(30 aa 이상)와 같은 복잡한 구간에서도 동일한 솔리톤 조합이 적용 가능함을 실험적으로 확인하였다.
이 접근법의 장점은 파라미터 수가 매우 적다는 점이다. 전통적인 구조 비교에서는 수천 개의 원자 좌표가 필요하지만, 솔리톤 모델은 각 구간당 3~4개의 실수 파라미터만으로 전체 형태를 기술한다. 따라서 대규모 단백질 데이터베이스에 대한 빠른 검색, 구조 예측, 그리고 변이 효과 분석 등에 활용될 수 있다. 한편, 현재 모델은 주로 단일 체인, 비교적 작은 단백질에 초점을 맞추었으며, 다중 체인 복합체나 큰 도메인 간 인터페이스에 대한 확장은 추가 연구가 필요하다.
결론적으로, 이 논문은 비선형 물리학의 초월 함수를 단백질 구조에 적용함으로써, 계층적 생물학적 분류와 원자 수준 물리 모델 사이의 연결 고리를 제공한다. 이는 단백질 구조 예측, 설계, 그리고 기능-구조 관계 해석에 새로운 정량적 도구를 제공할 잠재력을 가진다.
댓글 및 학술 토론
Loading comments...
의견 남기기