그래프 이론 기반 동적 힘 분광 데이터 자동 분류 방법
초록
동적 힘 분광 실험에서 얻은 힘‑거리 곡선을 상관관계 네트워크로 변환하고, 그래프 스펙트럼 분석을 통해 유사한 곡선들을 모듈로 구분한다. 이 방법은 ssDNA‑ssDNA, 펩타이드‑RNA(히브리드), 펩타이드‑금 표면 등 다양한 시스템에서 서로 다른 결합 메커니즘과 실험 조건을 자동으로 식별한다.
상세 분석
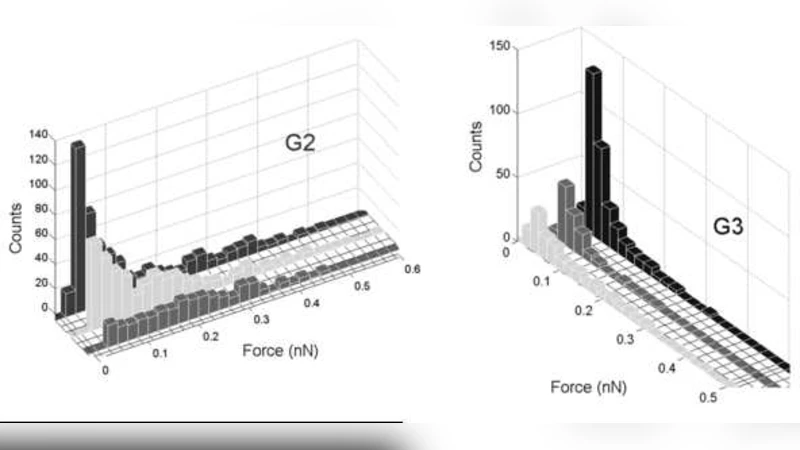
본 논문은 동적 힘 분광(Dynamic Force Spectroscopy, DFS) 실험에서 발생하는 높은 노이즈와 비특이 결합을 효과적으로 제거하기 위해 그래프 이론을 적용한 새로운 데이터 전처리 파이프라인을 제시한다. 먼저, 동일한 실험 조건 하에 반복적으로 얻어진 수천 개의 힘‑거리 곡선을 각각 하나의 노드로 매핑하고, 두 곡선 사이의 피어슨 상관계수를 가중치로 하는 완전 연결 그래프를 구축한다. 이때 상관계수 임계값을 조정하여 의미 있는 연결만을 남기고, 희소 행렬 형태의 인접 행렬을 얻는다. 구축된 네트워크는 비선형 차원 축소와 스펙트럴 클러스터링을 통해 토폴로지적 모듈(community)로 분할된다. 구체적으로 라플라시안 행렬의 고유벡터를 이용한 Fiedler 벡터 분석과 모듈러리티 최적화를 결합함으로써, 서로 높은 상관성을 보이는 곡선들이 동일 모듈에 속하도록 자동 군집화한다.
실험적으로는 세 가지 바이오분자 시스템을 대상으로 검증하였다. 첫 번째는 상보적인 ssDNA‑ssDNA 결합으로, 단일 결합 이벤트와 다중 결합 이벤트가 명확히 구분되는 두 개의 모듈이 도출되었다. 두 번째는 HIV‑1 관련 펩타이드와 RNA 사이의 상호작용으로, 결합 위치와 당단백질 구조에 따라 세 개 이상의 서브모듈이 형성되었으며, 이는 기존의 단일 파라미터(예: rupture force) 기반 분석으로는 식별하기 어려운 미세한 차이를 포착한다. 세 번째는 펩타이드와 금 표면 간의 비특이 흡착 현상으로, 속도 의존성에 따라 두 개의 주요 모듈이 나타났으며, 빠른 풀링 속도에서는 비특이 결합이 우세함을 확인하였다.
이러한 결과는 그래프 기반 접근법이 전통적인 히스토그램 분석이나 단순 임계값 필터링보다 높은 민감도와 선택성을 제공함을 시사한다. 특히, 다중 결합 부위가 존재하거나 복합체의 구조적 이질성이 큰 시스템에서, 각 결합 경로별로 독립적인 통계적 특성을 추출할 수 있다. 또한, 라플라시안 스펙트럼을 이용한 모듈 탐지는 실험 파라미터(예: 풀링 속도, 온도, 탐침 간 거리)의 연속적인 변화를 연속적인 네트워크 변형으로 해석할 수 있게 하여, 실험 설계 단계에서 최적 조건을 사전 예측하는 데 활용 가능하다.
한계점으로는 상관계수 임계값 선택이 결과에 민감하게 작용한다는 점과, 매우 큰 데이터셋(수만 개 이상)에서는 그래프 구축 및 스펙트럴 분해에 고성능 컴퓨팅 자원이 필요하다는 점을 들 수 있다. 향후 연구에서는 커널 기반 유사도 측정이나 비선형 차원 축소 기법을 결합해 네트워크의 희소성을 더욱 강화하고, 실시간 데이터 스트리밍 환경에서 온라인 클러스터링을 구현하는 방안을 모색할 수 있다.
요약하면, 본 연구는 DFS 데이터의 복잡성을 그래프 이론이라는 수학적 틀로 재구성함으로써, 실험적 변동성과 비특이 결합을 자동으로 구분하고, 분자 상호작용의 미세한 차이를 정량화할 수 있는 강력한 도구를 제공한다.
댓글 및 학술 토론
Loading comments...
의견 남기기