베이지안 신경망으로 복합 질환 유전 연관 분석 혁신
본 논문은 대규모 유전체 연관 연구에서 유전형‑표현형 관계를 모델링하기 위해 베이지안 신경망(BNN)을 제안한다. 자동 관련성 판단(ARD) 사전과 Hamiltonian Monte Carlo(HMC) 샘플링을 결합해 변수 중요도를 불확실성까지 제공하며, GPU 가속을 통해 수백만 SNP 데이터도 몇 시간 내에 분석한다. 시뮬레이션과 실제 GWAS 데이터에서 기존 MDR, BEAM, 랜덤 포레스트 등에 비해 높은 검출력과 효율성을 입증한다.
저자: Andrew L. Beam, Alison Motsinger-Reif, Jon Doyle
본 연구는 복합 질환의 유전적 원인을 밝히기 위해 대규모 유전체 연관 연구(GWAS)에서 발생하는 계산적·통계적 난제를 해결하고자 한다. 전통적인 방법인 MDR, BEAM, 랜덤 포레스트 등은 마커 수가 급증함에 따라 조합 탐색 비용이 기하급수적으로 늘어나거나, MCMC 샘플링이 차원에 비례해 비효율적이다. 이를 극복하기 위해 저자들은 베이지안 신경망(BNN)을 제안한다.
BNN은 다층 퍼셉트론 구조를 기반으로 하며, 입력 변수(각 SNP)와 은닉 유닛 사이의 가중치를 ARD(Automatic Relevance Determination) 사전으로 묶는다. ARD 사전은 동일 입력에 연결된 모든 가중치를 평균 0, 공통 분산 σ²_j를 갖는 정규분포로 가정하고, σ²_j 자체는 역감마 사전으로 설정한다. 이렇게 하면 사후에서 σ²_j가 작으면 해당 SNP이 모델에 거의 기여하지 않음을 의미하고, 큰 σ²_j는 중요한 변수를 나타낸다.
베이지안 추론을 위해 저자들은 Hamiltonian Monte Carlo(HMC)를 채택한다. HMC는 로그 사후의 기울기를 이용해 물리적 시스템의 운동 방정식을 시뮬레이션함으로써 고차원 공간에서도 효율적인 탐색이 가능하다. 구체적으로, 매 반복마다 (1) ARD 하이퍼파라미터 σ²_j를 Gibbs 샘플링으로 업데이트하고, (2) 고정된 σ²_j 하에서 가중치 θ를 HMC로 샘플링한다. 이 두 단계가 교대로 수행돼 전체 사후 분포를 정확히 추정한다.
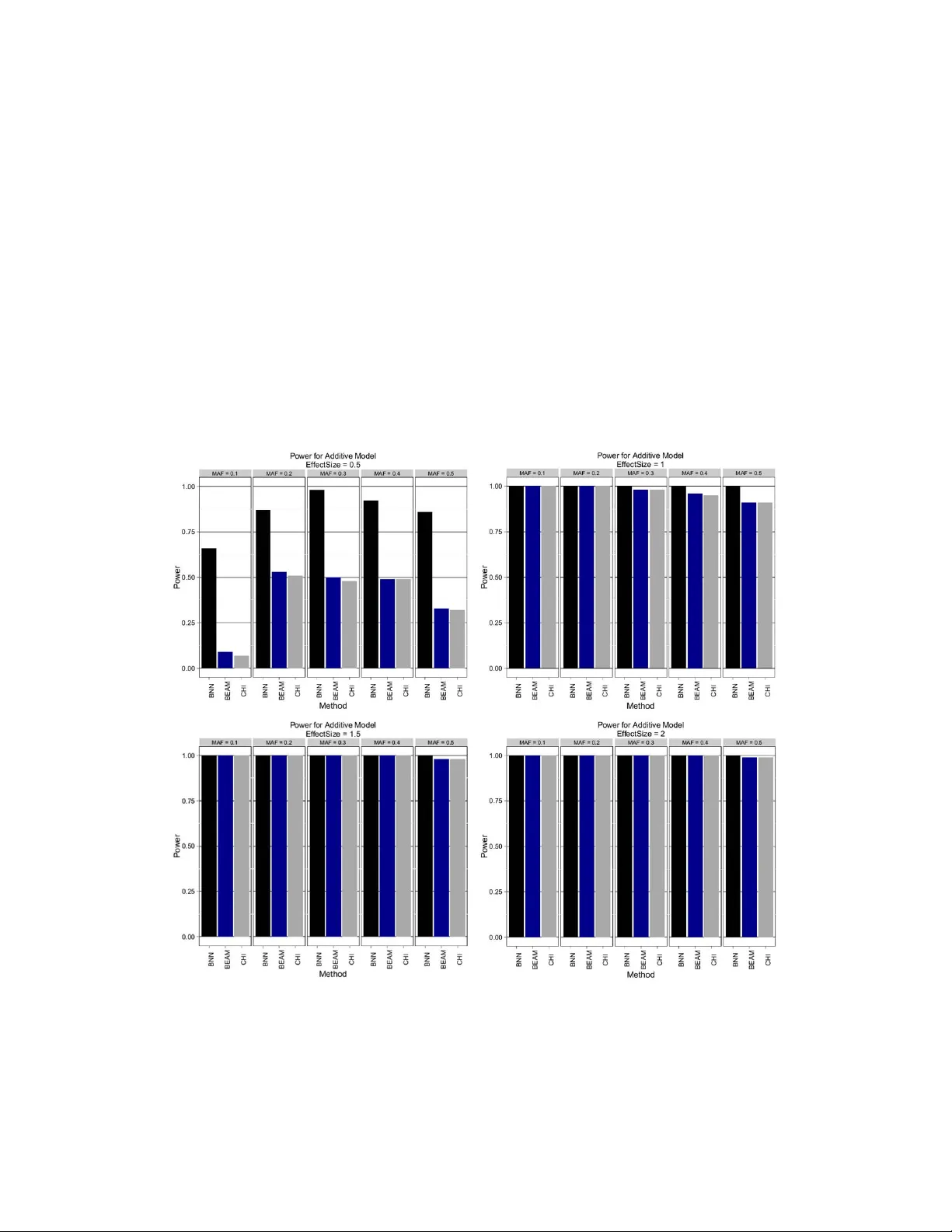
계산 효율성을 높이기 위해 저자들은 GPU 기반 병렬 구현을 도입했다. 그래디언트 계산과 행렬 연산을 GPU에서 수행함으로써 전통적인 CPU 구현 대비 수십 배에서 수백 배까지 속도가 향상되었다. 실험에서는 시뮬레이션 데이터(단일 마커 효과, 2‑way·3‑way 상호작용)와 실제 프리온병 GWAS 데이터를 사용했다. BNN은 모든 경우에서 높은 정확도와 AUC를 기록했으며, 특히 고차원 상호작용을 탐지할 때 조합을 전부 열거하지 않아도 효과적인 신호 포착이 가능했다. 변수 중요도는 사후 σ²_j 분포를 통해 직접 해석 가능했으며, 이는 퍼뮤테이션 기반 p‑값 계산을 대체한다.
비교 대상인 MDR는 전통적인 전수 탐색으로 인해 계산 시간이 급증했고, BEAM은 MCMC 반복 수가 SNP 수의 제곱에 비례해 비현실적인 수준이었다. 랜덤 포레스트와 부스팅 트리는 변수 중요도는 제공하지만 불확실성 추정이 부족했다. 반면 BNN은 변수 중요도와 그 불확실성을 동시에 제공하면서도 GPU 가속으로 실용적인 분석 시간을 확보했다.
결론적으로, 베이지안 신경망은 (1) 복잡한 비선형 및 상호작용 효과를 자동으로 모델링, (2) 변수 선택과 불확실성 추정을 통합, (3) GPU 가속을 통한 대규모 데이터 처리 능력이라는 세 가지 핵심 장점을 갖는다. 이는 현재와 미래의 대규모 GWAS 및 전장 유전체 시퀀싱 연구에 적용 가능하며, 유전적 위험 요인 탐색의 새로운 표준이 될 잠재력을 보여준다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기