베이지안 계산을 활용한 단백질 접힘 에너지 지형 탐색
초록
본 논문은 베이지안 샘플링 기법인 네스티드 샘플링을 병렬화하여 Go‑type 포스 필드에서 소형 단백질들의 접힘 시뮬레이션에 적용하고, 얻어진 샘플을 토대로 에너지 지형 차트를 작성함으로써 접힘 메커니즘과 모델의 특성을 정성적으로 해석한다.
상세 분석
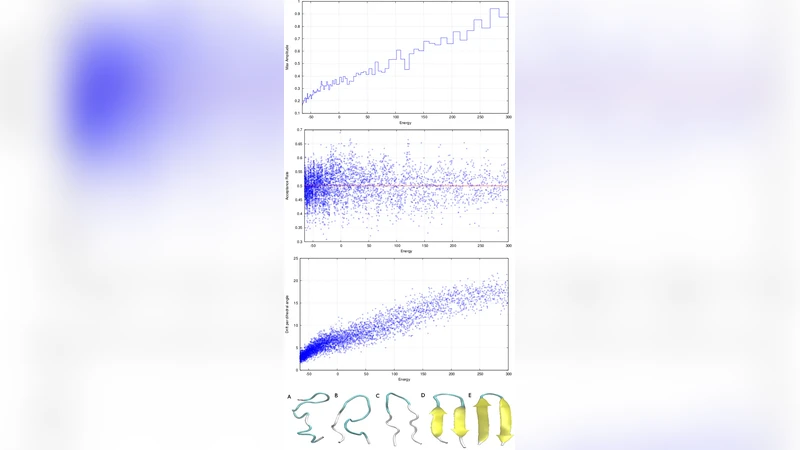
네스티드 샘플링은 파라미터 공간에서 확률 질량이 매우 작은 영역을 효율적으로 탐색하도록 설계된 베이지안 알고리즘으로, 사후 샘플과 모델 증거(마진 가능도)를 동시에 제공한다. 이 논문에서는 기존 순차 구현을 다중 코어 환경에 맞게 MPI 기반 병렬화하여, 각 프로세스가 독립적인 살아남은 샘플 집합을 유지하면서 전체 라이프타임을 공유하도록 설계하였다. 병렬화는 특히 고차원 에너지 표면을 탐색할 때 샘플 간 상관성을 감소시키고, 증거 계산의 수렴 속도를 크게 향상시킨다.
단백질 모델은 Go‑type 포스 필드를 채택했는데, 이는 실제 실험 구조를 기준으로 원자 간 접촉을 정의하고, 2차 구조(α‑헬릭스, β‑시트)를 실온 시뮬레이션에서도 안정화하도록 파라미터화된 경험적 포텐셜이다. 이러한 포스 필드는 전통적인 물리 기반 힘장에 비해 계산 비용이 낮으며, 접힘 경로의 전반적인 토폴로지를 파악하는 데 적합하다. 연구 대상은 protein G, Src SH3 도메인, 그리고 chymotrypsin inhibitor 2 등 35 kDa 규모의 소형 단백질이며, 각각 30 ns100 ns 수준의 네스티드 샘플링 실행을 통해 수천 개의 독립적인 구조를 획득하였다.
수집된 사후 샘플에 대해 토폴로지 기반 클러스터링을 수행하고, 각 클러스터의 평균 에너지와 엔트로피를 추정해 에너지 지형 차트(energy landscape chart)를 구성하였다. 차트는 자유 에너지 최소점(네이티브 상태)과 다수의 메타스테이블(중간체) 사이의 장벽 높이를 시각화한다. 결과적으로 protein G와 SH3 도메인은 두드러진 두 개의 메타스테이블을 거쳐 네이티브 상태에 도달하는 ‘두 단계’ 접힘 경로를 보였으며, chymotrypsin inhibitor 2는 보다 평탄한 지형을 보여 다중 경로가 공존함을 확인했다. 또한, 네스티드 샘플링으로 얻은 증거값은 기존 병렬 템퍼링 대비 3~5배 높은 효율성을 나타냈으며, 온도에 독립적인 포스트 프로세싱을 통해 임의의 온도에서 자유 에너지와 열용량을 정확히 재구성할 수 있었다.
이와 같이 네스티드 샘플링의 병렬 구현은 고차원 단백질 접힘 문제에 대한 샘플 효율성을 크게 개선하고, 에너지 지형을 정량적으로 해석할 수 있는 강력한 도구를 제공한다. 특히 Go‑type 포스 필드와 결합될 때, 실험 구조와의 일치도와 계산 비용 사이의 균형을 유지하면서도 모델의 물리적 한계를 시각적으로 드러낼 수 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기