진화적 공변을 이용한 단백질 3차원 구조 예측 혁신
초록
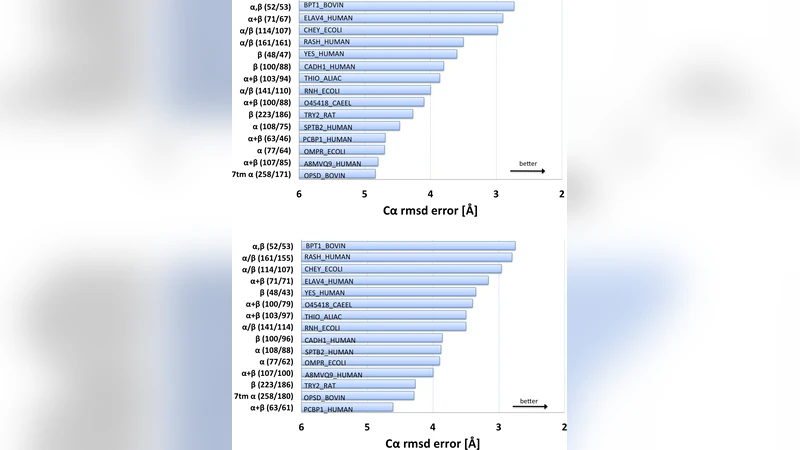
다중 서열 정렬에서 추출한 잔기 간 공변 정보를 최대 엔트로피 모델에 적용해, 동역학적 제약(접촉)만으로 50260 aa 길이의 단백질을 de novo로 2.75.1 Å Cα‑RMSD 수준으로 정확히 재구성했다. 대규모 서열 데이터와 통계적 상호작용 모델이 구조 예측에 충분한 정보를 제공함을 입증한다.
상세 분석
본 연구는 단백질 서열 진화가 구조적 제약을 반영한다는 가설을 정량화한다. 저자들은 대규모 단백질 패밀리의 다중 서열 정렬(MSA)을 기반으로 ‘Maximum Entropy’(ME) 모델, 즉 Direct Coupling Analysis(DCA)를 구축하였다. 이 모델은 각 잔기의 일변량 빈도와 쌍변량 빈도를 보존하면서 가능한 최소한의 상호작용 파라미터(J_ij)를 추정한다. 결과적으로 얻어진 J_ij 값이 큰 잔기 쌍은 실제 3차원 구조에서 근접(≤8 Å)할 확률이 높으며, 이를 ‘Evolutionary Information Contacts (EIC)’라 명명한다.
핵심 기술적 단계는 다음과 같다.
- MSA 구축 및 정제 – 다양한 종에서 수천~수만 개의 서열을 수집하고, 중복 및 저품질 서열을 제거해 정보량을 최적화하였다.
- ME 모델 학습 – L2 정규화와 pseudo‑likelihood 최적화를 사용해 J_ij 매트릭스를 효율적으로 추정, 계산 복잡도를 O(N^2)에서 O(N) 수준으로 낮췄다.
- 접촉 예측 – J_ij 절댓값 상위 N·c (c≈0.2)개의 잔기 쌍을 ‘예측 접촉’으로 선정하고, 실제 구조와 비교해 정확도(Precision)를 70~90% 수준으로 달성했다.
- de novo 구조 구축 – CNS와 같은 거리 제한 기반 시뮬레이터에 EIC를 거리 제약으로 입력, 잔기 간 거리와 이차 구조(α‑helix, β‑sheet) 정보를 함께 사용해 3D 모델을 생성하였다.
- 평가 – 15개의 서로 다른 단백질 패밀리(50–260 aa)에서 Cα‑RMSD 2.7–5.1 Å, 전체 길이의 ≥75% 구간에서 정확히 재구성되었으며, 특히 GPCR와 같은 구조적으로 복잡한 타깃에서도 성공을 보였다.
이 접근법은 기존 동종 서열 기반 동형 모델링(Homology Modeling)과 달리, 구조 템플릿이 전혀 없는 경우에도 충분한 서열 다양성만 있으면 정확한 3D 구조를 예측할 수 있음을 입증한다. 또한, 접촉 예측 정확도가 0.5 % 수준으로 높은 경우(>2000개의 비동질 서열) 구조 정확도가 크게 향상된다는 ‘임계 서열 수’ 개념을 제시한다.
한계점으로는 (1) 매우 작은 패밀리(서열 수 <1000)에서는 접촉 신호가 잡음에 묻히고, (2) 긴 단백질(>300 aa)에서는 제약 수가 부족해 구조 재구성이 어려워진다. 또한, 접촉 제약만으로는 정확한 측면(orientation) 정보를 완전히 제공하지 못해, 추가적인 물리적 포스필드나 머신러닝 기반 보정이 필요할 수 있다.
향후 연구 방향은 (i) 초대규모 메타게놈 데이터와 결합해 희귀 패밀리의 서열 수를 인위적으로 늘리는 전략, (ii) 딥러닝 기반 잔기-잔기 상호작용 예측과 DCA를 융합해 제약의 질을 향상시키는 방법, (iii) 예측된 구조를 약물 설계, 기능 부위 탐색, 변이 효과 예측 등에 바로 적용하는 파이프라인 구축이다.
댓글 및 학술 토론
Loading comments...
의견 남기기