복잡망 기반 유전체 서열 분류를 위한 혁신적 특징 추출 기법
초록
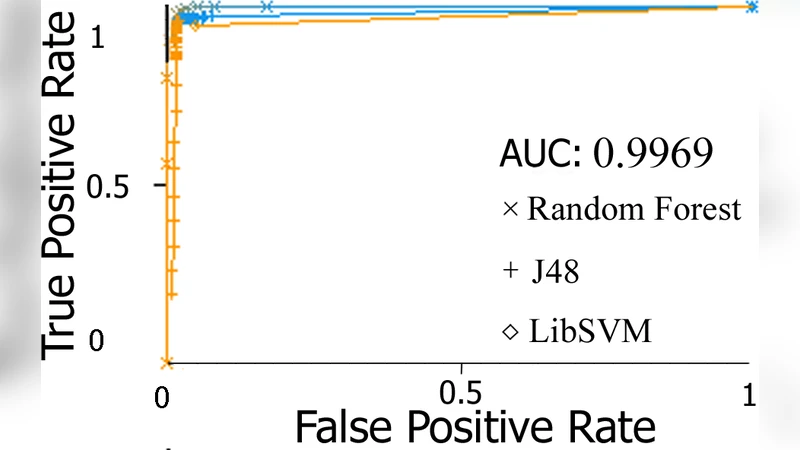
본 연구는 염기·다이뉴클레오티드·트리뉴클레오티드 수준에서 복잡망 구조와 엔트로피를 결합한 특징 벡터를 생성하고, 이를 이용해 코딩, 인터제닉, 전사 시작 부위(TSS) 서열을 분류한다. 랜덤 포레스트가 91.2%의 최고 정확도를 기록하였다.
상세 분석
논문은 유전체 서열을 세 가지 스케일(1‑mer, 2‑mer, 3‑mer)로 변환한 뒤, 각각을 노드로 하는 복잡망을 구축한다. 노드 연결은 ‘워드 사이즈(WS)’와 ‘스텝(P)’ 파라미터에 따라 순차적으로 읽은 k‑mer 사이의 인접성을 기반으로 정의되며, WS∈{1,2,3}, P∈{1,2,3} 조합으로 총 6개의 비방향망을 생성한다. 각 망에 대해 평균 최단 경로 길이, 클러스터링 계수, 다양한 중심성(정도, 근접성, 매개성, 효율성), 평균 차수, 모티프 빈도, 커뮤니티 수 등 전통적인 복잡망 지표와 노드 차수의 평균·표준편차·최대·최소값을 추출한다. 동시에 정보이론적 특성으로 전체 서열, 2‑mer, 3‑mer에 대한 엔트로피, 엔트로피 합, 최대 엔트로피를 계산한다. 이렇게 얻어진 복합 특징들은 하나의 고차원 벡터로 결합되어 각 서열을 대표한다. 특징 선택 없이 모든 변수를 그대로 사용해 WEKA의 6가지 분류기(Random Forest, J48, SVM, MLP, IBK, Naive Bayes)를 적용했으며, 10‑fold 교차검증 결과 Random Forest가 91.2%, J48이 89.1%, SVM이 84.8%의 정확도를 보였다. ROC 곡선에서도 세 클래스(코딩, 인터제닉, hspromoter) 모두 높은 AUC를 기록, 특히 인터제닉 영역은 높은 염기 반복성으로 인해 네트워크 구조적 차이가 뚜렷하게 드러났다. 연구는 파라미터 조합을 확대하면 망의 수와 특징이 증가해 분류 성능이 더욱 향상될 가능성을 제시하고, miRNA, 전사체, 전사 조절 서열 등 다양한 유전체 요소에 적용할 여지를 남긴다.
댓글 및 학술 토론
Loading comments...
의견 남기기