단백질 서열 탐색을 가속화하는 펩타이드 워드 라티스 기반 DRIP 개선
본 논문은 기존 동적 베이지안 네트워크 기반 펩타이드 식별 도구 DRIP에 두 가지 주요 개선을 제안한다. 첫째, 자연어 처리에서 활용되는 워드 라티스(word lattice)를 도입해 후보 펩타이드 집합을 하나의 그래프 구조로 압축함으로써 중복 연산을 공유하고, 실험에서 10배 이상 속도 향상을 달성하였다. 둘째, 최대우도 추정이 아닌 최대상호정보(maximum mutual information) 추정을 이용한 판별적 학습(discrimina…
저자: Shengjie Wang, John T. Halloran, Jeff A. Bilmes
본 연구는 대규모 shotgun proteomics 실험에서 수만 개에 달하는 tandem mass 스펙트럼을 빠르고 정확하게 펩타이드에 매핑하는 문제를 다룬다. 기존 DRIP(Dynamic Bayesian Network for Rapid Identification of Peptides)은 동적 베이지안 네트워크를 이용해 관측 스펙트럼과 이론 스펙트럼을 정렬하고, Viterbi 알고리즘으로 최적 경로를 찾아 점수를 산출한다. 그러나 후보 펩타이드 집합을 개별적으로 스코어링하는 전통적인 방식은 후보 수가 많을수록 연산 비용이 급증한다는 한계가 있었다.
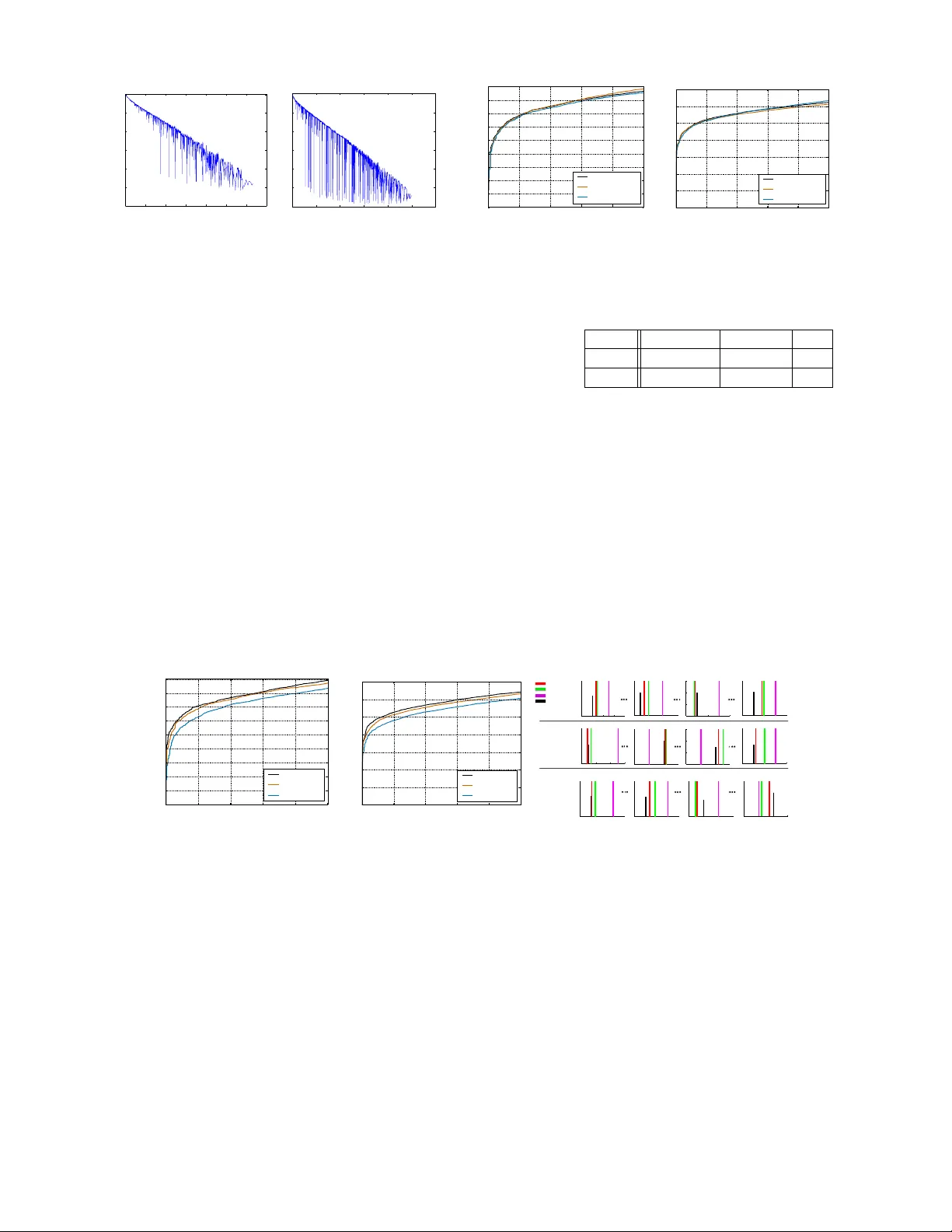
이를 해결하기 위해 논문은 두 가지 주요 개선을 제안한다. 첫 번째는 워드 라티스(word lattice)를 도입하는 것이다. 라티스는 문자열 집합을 공유 접두·접미를 이용해 하나의 유향 그래프로 압축하는 자료구조로, 자연어 처리와 음성 인식 분야에서 후보 집합을 효율적으로 탐색하는 데 널리 사용된다. 펩타이드 후보 집합을 라티스로 표현하면, 동일한 아미노산 서열 조각을 공유하는 여러 후보가 같은 그래프 경로를 따라 이동하게 된다. Viterbi 디코딩 단계에서 이론 피크에 대한 가우시안 점수 계산을 한 번만 수행하고, 해당 점수를 라티스 상의 모든 경로에 재사용함으로써 중복 연산을 크게 줄일 수 있다. 실험 결과, yeast와 worm 데이터셋에서 라티스 기반 DRIP은 연산량을 85 %~93 % 감소시켰으며, 전체 실행 시간은 10배 이상 단축되었다.
두 번째 개선은 학습 목표 함수를 최대우도(maximum likelihood)에서 최대상호정보(maximum mutual information, MMI)로 전환한 판별적 학습(discriminative training)이다. 기존 DRIP은 고신뢰 PSM(펩타이드‑스펙트럼 매치)만을 사용해 모델 파라미터를 추정했지만, 실제 식별 성능은 정답과 오답을 구분하는 능력에 달려 있다. 논문은 양성 샘플(고신뢰 PSM)과 음성 샘플(동일 전구체 질량 범위 내 무작위 후보) 사이의 로그우도 차이를 최대화하도록 파라미터를 최적화한다. 이 방식은 삽입·삭제 확률과 가우시안 파라미터를 보다 효과적으로 조정하게 하여, 1 % FDR 조건에서 식별된 스펙트럼 수를 평균 5 %~12 % 증가시켰다.
또한 전하 상태가 서로 다른 스펙트럼 간 점수 비교가 어려운 문제를 해결하기 위해 전하별 보정(calibration) 절차를 도입하였다. 새로운 디코이(peptide decoy) 집합 D_dd를 생성하고, 각 전하별 디코이 점수를 정규화하여 목표 펩타이드와 디코이 점수를 동일한
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기