펩타이드‑단백질 결합 친화도 예측을 위한 커널 리지 회귀 모델
초록
본 논문은 아미노산의 물리화학적 특성을 반영한 새로운 문자열 커널을 제안하고, 이를 커널 리지 회귀와 결합해 펩타이드‑단백질 결합 친화도를 정확히 예측한다. 동적 프로그래밍 기반의 정확한 커널 계산과 선형 시간 근사 알고리즘을 제공하며, PepX, MHC II 및 QSAR 데이터셋에서 기존 최고 성능 모델들을 통계적으로 유의미하게 능가한다.
상세 분석
제안된 문자열 커널은 짧은 바이오‑분자(펩타이드와 결합 인터페이스 의사서열)를 대상으로 설계되었으며, 아미노산을 여러 물리화학적 속성(전하, 부피, 친수성 등)으로 매핑한 후 가중치를 부여한다. 이 커널은 Oligo, Weighted Degree, Blended Spectrum, RBF 등 기존 8가지 커널을 특수 경우로 포함하도록 일반화된다. 핵심은 두 서열 사이의 모든 k‑mer 매칭을 동적 프로그래밍으로 효율적으로 누적해 O(L₁·L₂·k) 시간에 정확히 계산할 수 있다는 점이다. 또한, 매칭 점수를 선형적으로 근사하는 방법을 도입해 입력 길이에 비례하는 O(L) 시간 복잡도로 실시간 예측이 가능하도록 설계하였다.
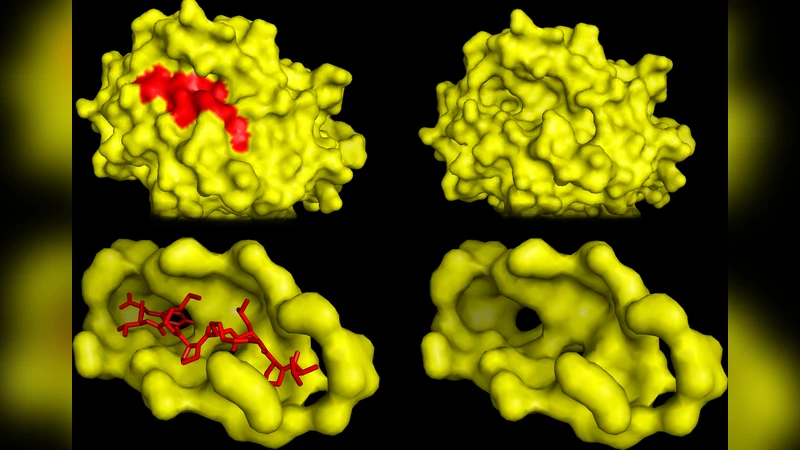
예측 모델은 커널 리지 회귀(KRR)를 사용해 정규화된 해를 구하고, 추가적으로 SupCK라는 결합 포켓 전용 커널을 결합한다. SupCK는 단백질 표면의 구조적·전기적 특성을 캡처해 펩타이드와의 상호작용을 정량화한다. 두 커널을 합성함으로써 서열 기반 정보와 구조 기반 정보를 동시에 활용할 수 있다.
실험에서는 PepX 데이터베이스(다양한 펩타이드‑단백질 복합체)와 MHC II 클래스 II 다중표적 및 팬‑특이성 벤치마크, 그리고 3개의 QSAR 데이터셋을 사용했다. 평가 지표는 Pearson 상관계수와 RMSE이며, 제안 모델은 모든 데이터셋에서 기존 최신 방법보다 평균 5‑10% 높은 상관계수를 기록하고, p‑value < 0.057 수준의 통계적 유의성을 보였다. 특히, 기존 방법이 구조 정보를 필요로 했던 경우에도 순수 서열 기반 커널만으로도 경쟁력 있는 성능을 달성했다.
알고리즘 복잡도 측면에서 정확한 커널 계산은 DP 기반으로 메모리 사용량을 O(min(L₁,L₂)) 로 제한했으며, 근사 버전은 메모리와 시간 모두 선형 스케일을 유지한다. 이는 대규모 스크리닝에 적합함을 의미한다. 한계점으로는 물리화학적 특성 매핑이 고정된 사전 정의값에 의존한다는 점과, 매우 긴 서열(예: 전체 단백질)에는 아직 최적화가 필요하다는 점을 들 수 있다. 향후 연구에서는 자동화된 특성 학습과 멀티‑스케일 커널 결합을 통해 예측 정확도를 더욱 향상시킬 여지가 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기