메타게놈 읽기 압축을 위한 MCUIUC 프레임워크
초록
본 논문은 메타게놈 데이터의 대용량 저장 문제를 해결하기 위해, 읽기들을 미생물 종별 고유 식별자로 분류하고, 각 종에 대해 참조 기반 정렬 및 미정류(reads) 집합에 대한 어셈블리를 수행한 뒤, 위치 인코딩을 이용한 무손실 압축을 적용하는 MCUIUC 알고리즘을 제안한다. 15종의 합성 데이터를 대상으로 실험했으며, 향후 정확도와 속도 개선 방안을 제시한다.
상세 분석
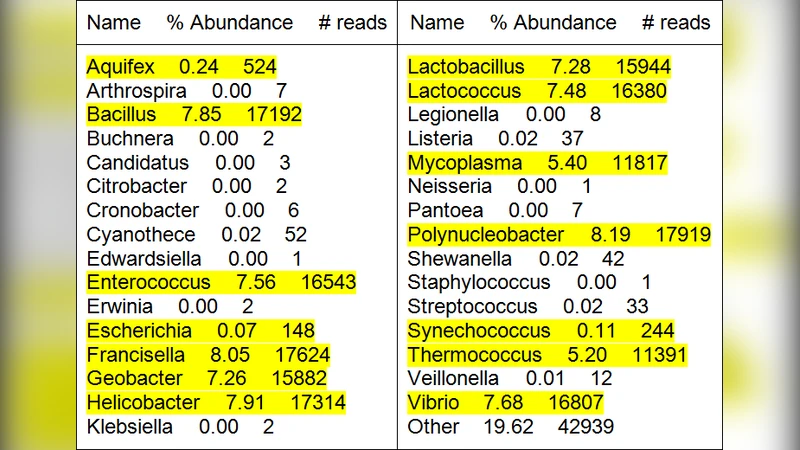
MCUIUC는 메타게놈 압축을 위한 최초의 체계적 파이프라인으로, 크게 세 단계로 구성된다. 첫 번째 단계는 Metaphyler를 이용한 ‘거친’ 종(Genus) 수준 식별이다. Metaphyler는 20 bp 이상의 마커 서열이 종 고유임을 전제로, 메타데이터에서 마커를 스캔해 가능한 속(genus) 목록과 그 풍부도(abundance)를 출력한다. 이때 false‑positive와 미식별 종이 존재하므로, 논문에서는 마커 수, 게놈 길이, 예상 풍부도 등을 종합해 최종 후보 속을 선정한다. 두 번째 단계는 후보 속에 속한 모든 종의 완전 게놈을 대상으로 Bowtie2를 이용해 초고속 정렬을 수행하고, 정렬된 읽기를 속별로 집합화한다. 각 속 내에서 가장 많은 읽기가 매핑된 종을 ‘대표(reference) 종’으로 정의하고, 해당 대표 게놈에 대해 위치와 변이 정보를 Golomb‑code 기반의 포지셔널 인코딩으로 압축한다. 정렬에 실패한 읽기(unaligned reads)는 비율이 20~25 % 수준에 머무르므로, IDBA‑UD와 같은 메타어셈블러로 컨티그를 생성하고, BLAST을 통해 잠재적 출처를 추정한다. 추정된 출처가 새로운 속에 속하면 위 과정을 재귀적으로 적용한다. 마지막 단계에서는 속별 SAM 파일을 BAM으로 변환하고, CRAM 툴킷을 이용해 대표 게놈 대비 무손실 압축을 수행한다. 압축되지 않은 읽기와 메타데이터는 bzip2 등 표준 압축기로 포장한다.
핵심 기술적 인사이트는 (1) 메타게놈 데이터의 복합성을 ‘속 수준’으로 먼저 단순화함으로써 전체 정렬 비용을 크게 감소시킨 점, (2) 대표 게놈을 선택해 단일‑게놈 압축 알고리즘을 그대로 활용함으로써 기존 압축기술(예: DNACompress, CRAM)의 성능을 그대로 이식한 점, (3) 미정렬 읽기에 대해 어셈블리 후 컨티그를 새로운 참조로 재활용함으로써 압축 효율을 추가로 높인 점이다. 또한, 논문은 메타게놈 샘플이 600 GB를 초과하는 현실적인 규모에서도 파이프라인이 병렬화 가능함을 시사한다. 한계점으로는 Metaphyler의 종 식별 정확도가 낮을 경우 잘못된 대표 종 선택으로 압축 효율이 저하될 수 있다는 점과, 어셈블리 단계가 여전히 메모리·CPU 집약적이라는 점을 들었다. 향후 연구에서는 딥러닝 기반 마커 탐색, 동적 대표 종 교체, 그리고 컨티그 기반 다중‑레퍼런스 압축을 통해 이러한 약점을 보완하고자 한다.
댓글 및 학술 토론
Loading comments...
의견 남기기