케미시프트: 확률적 화학이동 할당을 통한 NMR 기반 단백질 구조 예측 혁신
초록
**
본 논문은 Phaistos 프로그램에 통합된 Chemshift 모듈을 제안한다. 화학이동(NMR) 데이터를 확률적 베이즈 프레임워크로 처리해 자동 할당과 물리적 힘장 사이의 가중치를 자동으로 최적화한다. 부분 할당만으로도 할당 정확도를 향상시키며, 구조 예측 과정에서 데이터 손실 없이 편향을 최소화한다.
**
상세 분석
**
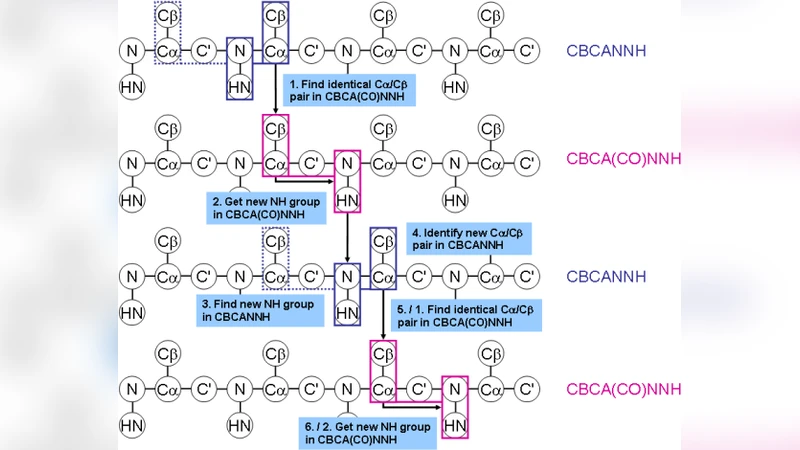
Chemshift 모듈은 기존 자동 할당 프로그램인 Autoassign와 FLYA가 갖는 한계를 정량적 베이즈 모델로 극복한다. 먼저, 화학이동의 측정값을 정규분포(평균 μ, 표준편차 σ)로 가정하고, 두 측정값 사이의 결합 확률을 σ²_i+σ²_j 를 이용한 가우시안 형태로 전개한다. 이는 중앙극한정리와 최대 엔트로피 원칙에 기반한 최소 편향 사전분포(log‑normal 혹은 normal)와 결합돼, nuisance 파라미터(예: 실험 오차, 스케일링)까지 동시에 샘플링한다.
Markov Chain Monte Carlo(MCMC)에서 Chemshift는 두 종류의 움직임을 정의한다. 하나는 “nuisance parameter moves”로, σ와 같은 실험 파라미터를 메트로폴리스 기준으로 업데이트한다. 다른 하나는 “assignment moves”로, 피크와 잔기의 매핑을 교환·재배치한다. 이러한 움직임은 각각의 사후 확률 비율에 의해 수용되며, 물리적 에너지(힘장)와 데이터 에너지(화학이동 차이)의 가중치 ω_data는 사전 정의가 아니라 MCMC 과정에서 자동으로 조정된다.
구조 예측 단계에서는 화학이동 예측 모델(SPARTA, SHIFTX 등)으로부터 얻은 μ_pred와 큰 σ_pred를 사용해 데이터 에너지 함수를 정의한다. 이때, 실제 측정값과 예측값 사이의 차이를 로그 σ + Δ²/(2σ²) 형태로 손실에 포함시켜, 데이터와 물리적 힘장의 균형을 정량화한다. 결과적으로, 부분 할당된 피크라도 구조가 변함에 따라 할당이 동적으로 재평가되므로, 초기 할당 오류가 구조 최적화 과정에서 자연스럽게 교정된다.
실험에서는 20~150 아미노산 길이의 소단백질을 대상으로 Autoassign와 FLYA와 비교했으며, Chemshift는 평균 96% 이상의 백본 화학이동 할당률과 0.4% 이하의 오류율을 기록했다. 특히, 피크 중복·노이즈·누락이 심한 경우에도 MCMC 기반 할당이 수렴하여, 전통적인 결정론적 할당이 실패하는 상황에서도 유의미한 구조 정보를 제공한다.
이 논문의 핵심 기여는 (1) 화학이동 할당을 확률적 베이즈 프레임워크에 통합, (2) 물리적 힘장과 실험 데이터의 가중치를 자동 최적화, (3) Phaistos와의 원활한 연동을 통한 구조 예측 파이프라인 구축이다. 향후 작업으로는 레퍼런스 오류 보정, 피크 강도 모델링, 그리고 NOE와 결합한 다중 데이터 융합이 제시된다.
**
댓글 및 학술 토론
Loading comments...
의견 남기기