Missing Heritability를 밝히는 교차 검증 기반 GWAS
본 연구는 5‑fold 교차 검증을 이용해 각 SNP의 예측력(R²)을 직접 측정하고, 예측력이 높은 상위 0.05% SNP 중 최대 5개를 전진 선택(stepwise)으로 조합하여 좁은 의미의 유전력(h²)을 추정한다. Arabidopsis 49가지 형질에 적용한 결과, 전체 유전체를 이용한 SNP‑BLUP보다 소수의 QTL가 더 큰 h²를 설명함을 보여, 기존 GWAS에서 놓친 ‘missing heritability’가 분석 방법의 부적절함…
저자: Xia Shen
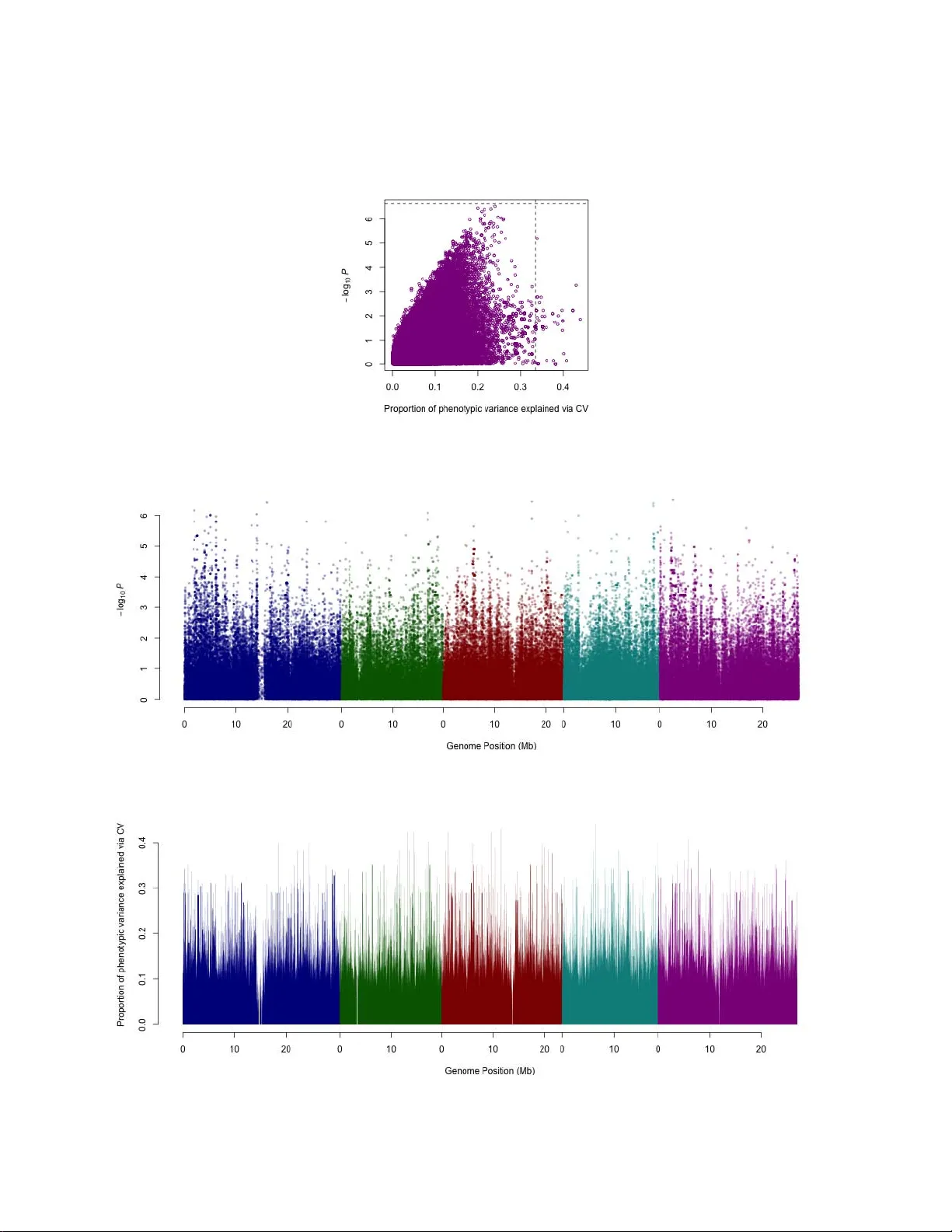
본 논문은 “cross‑validated genome‑wide association studies”(cvGWAS)라는 새로운 분석 프레임워크를 제시한다. 기존 GWAS는 각 마커에 대해 p‑값을 계산하고, 통계적 유의성을 기준으로 후보 SNP를 선정한다. 그러나 p‑값은 효과 크기와는 별개이며, 실제 형질 변이를 예측하는 능력을 반영하지 못한다. 저자는 이를 보완하기 위해 5‑fold 교차 검증을 이용해 각 SNP의 예측력(R²_SNP)을 직접 측정한다. 구체적으로, 전체 샘플을 5개의 겹치지 않는 테스트 집합(각 20%)과 훈련 집합(80%)으로 나누고, 훈련 집합에서 단일 SNP에 대한 선형 회귀 모델을 적합한다. 이후 테스트 집합에서 예측값을 얻고, 실제 형질값과의 상관계수 제곱을 구한다. 이 과정을 5번 반복한 뒤 평균 R²_SNP를 각 SNP의 ‘예측력’ 지표로 사용한다.
다음으로, 전체 216,130개의 SNP 중 상위 0.05% (≈108개)의 R²_SNP 값을 가진 SNP를 후보군으로 선정한다. 이 후보군에 대해 전진 선택(stepwise forward) 절차를 적용해 최대 5개의 SNP 조합을 찾는다. 선택 과정에서도 5‑fold CV를 사용해 조합의 예측력을 평가한다. 이렇게 선정된 QTL 집합이 설명하는 변이 비율을 h²_QTL 로 정의한다.
전체 마커를 이용한 전통적인 유전력 추정은 ‘SNP‑BLUP’(ridge regression) 모델을 사용한다. 이 모델을 동일한 5‑fold CV 절차에 적용해 얻은 변이 설명 비율을 h²_G 로 정의한다. 저자는 49가지 Arabidopsis 형질(꽃 개화, 발달, 방어, 이온omics) 각각에 대해 h²_QTL와 h²_G를 비교한다. 결과는 대부분의 형질에서 h²_QTL가 h²_G보다 크게 높으며, 특히 방어와 이온omics 형질은 매우 희소형(소수의 QTL) 구조를 보인다. 반면, 전형적인 개화 형질은 다소 다중유전적 특성을 띠지만, 여전히 2~5개의 QTL만으로 상당한 h²를 포착한다.
이러한 결과는 두 가지 중요한 시사점을 제공한다. 첫째, 전체 마커를 사용해 전체 유전력을 추정하는 방법은 효과 추정의 분산을 크게 증가시켜 실제 효과를 희석한다. 따라서 ‘전체 마커 기반 h²’는 실제 유전력을 과소평가한다는 것이 저자의 주장이다. 둘째, 예측력 기반으로 선별된 소수의 QTL는 실제로 형질 변이를 크게 설명할 수 있으며, 이는 missing heritability가 분석 방법의 부적절함에서 비롯된다는 새로운 관점을 제시한다.
방법론적 구현 측면에서, 저자는 R 패키지(cvGWAS, FWDselect, hglm, bigRR)를 공개했으며, 데이터와 분석 코드는 모두 공개되어 재현성을 확보했다. 또한, Supplementary Table 2에 각 형질별 선정된 QTL와 h²_QTL, h²_G 값이 상세히 제공된다.
하지만 몇 가지 제한점도 존재한다. 첫째, 5‑fold CV는 샘플 수가 제한된 경우(예: 84~194개) 변동성이 클 수 있다. 둘째, 선형 회귀 모델은 SNP 간 상호작용이나 비선형 효과를 포착하지 못한다. 셋째, Arabidopsis와 같은 모델 식물에서의 결과가 인간과 같은 복잡한 유전체에 그대로 적용될지는 추가 검증이 필요하다.
향후 연구에서는 (1) 더 많은 폴드 혹은 부트스트랩 기반의 안정적인 예측력 추정, (2) 비선형 모델(예: 랜덤 포레스트, 딥러닝)과의 결합, (3) 베이지안 프레임워크를 통한 효과 추정의 정밀도 향상 등을 통해 cvGWAS를 확장할 수 있다. 또한, 다중형질 분석을 통해 공통 QTL를 탐색하거나, 환경과의 상호작용을 고려한 모델링을 수행한다면, 복합 형질의 유전 구조를 보다 정밀하게 규명할 수 있을 것이다.
결론적으로, 본 연구는 ‘예측력 기반 마커 선택’이라는 새로운 관점을 도입함으로써, 기존 GWAS가 놓친 유전력을 회복하고, 복잡 형질의 유전적 아키텍처를 보다 현실적으로 파악할 수 있음을 보여준다. 이는 유전체 예측, 육종, 인간 질병 연구 등 다양한 분야에 중요한 방법론적 전환을 제시한다.
원본 논문
고화질 논문을 불러오는 중입니다...
댓글 및 학술 토론
Loading comments...
의견 남기기