시퀀스 데이터 정확·고속 분할을 위한 Segmentor3IsBack 패키지
초록
본 논문은 RNA‑Seq와 같은 차세대 시퀀싱 데이터의 유전자 경계 탐지를 위해, 기존 CGH 배열 분석에 성공한 Pruned Dynamic Programming(PDP) 알고리즘을 음이항 분포에 맞게 확장한다. 분산 파라미터를 사전 추정하고, 오라클 페널티를 이용해 최적 세그먼트 수를 선택한다. 구현된 R 패키지는 CRAN에 공개되어 실제 데이터셋에서 높은 정확도와 거의 선형 시간 복잡도를 보이며, 코딩·비코딩 영역 구분에 유용함을 입증한다.
상세 분석
본 연구는 차세대 시퀀싱(NGS) 데이터, 특히 RNA‑Seq와 같은 카운트 기반 데이터의 구간(segmentation) 문제를 해결하기 위해, 기존에 CGH(array comparative genomic hybridization) 데이터에서 성공적으로 적용된 Pruned Dynamic Programming(PDP) 알고리즘을 음이항(Negative Binomial) 모델에 맞게 재구성하였다. 음이항 분포는 과분산(over‑dispersion)을 자연스럽게 설명할 수 있어, RNA‑Seq와 같은 고분산 카운트 데이터에 적합하다. 논문은 먼저 분산 파라미터(φ)를 사전 추정하는 절차를 제시한다. 구간별 평균 μ를 최대우도 추정하고, 전체 데이터에 대해 φ를 모멘트 방법 혹은 베이지안 사전으로 추정함으로써, PDP 알고리즘이 요구하는 고정된 분산값을 제공한다.
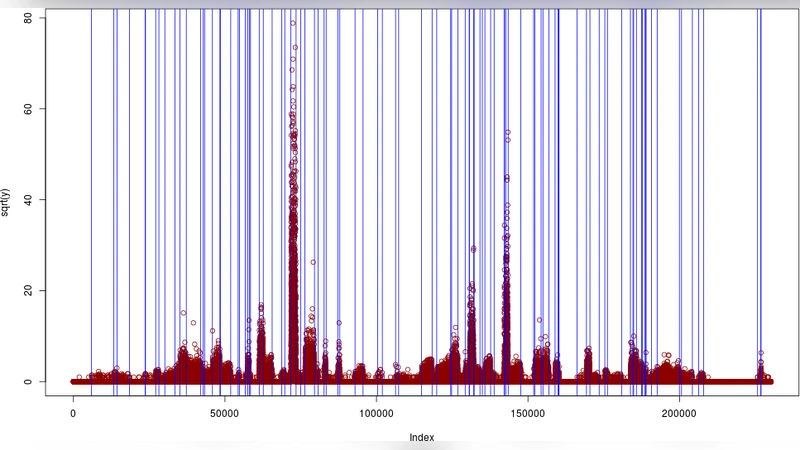
PDP 알고리즘 자체는 동적 계획법(DP)의 탐색 공간을 ‘가지치기(pruning)’ 함으로써, 전통적인 O(n²) 복잡도를 O(n·K) 수준으로 낮춘다. 여기서 n은 데이터 포인트 수, K는 최종 세그먼트 수이다. 알고리즘은 각 후보 구간에 대해 손실 함수(음이항 로그우도)의 누적값을 계산하고, 현재까지 최적이라고 판단되는 경로만을 유지한다. 이 과정에서 ‘convexity’와 ‘monotonicity’ 속성을 활용해 불필요한 후보를 빠르게 제거한다. 결과적으로 수십만 개의 카운트 데이터를 수 초 내에 정확히 분할할 수 있다.
세그먼트 수 K의 선택은 모델 복잡도와 과적합 사이의 균형을 맞추는 핵심 문제이다. 저자들은 ‘오라클 페널티(oracle penalty)’ 프레임워크를 도입해, 데이터에 내재된 잡음 수준과 분산 추정치를 반영한 적절한 페널티 함수를 설계한다. 구체적으로, BIC와 유사한 형태의 페널티 λ·K를 사용하되, λ를 φ와 샘플 크기 n에 의존하도록 조정한다. 이를 통해 실제 데이터에 적용했을 때, 과소/과다 분할을 방지하고 생물학적으로 의미 있는 구간을 도출한다.
패키지 구현 측면에서는 R 언어와 C++(Rcpp) 기반의 고성능 코드를 결합해, 사용자가 친숙한 R 인터페이스를 통해 손쉽게 파라미터 설정, 결과 시각화, 그리고 downstream 분석(예: 차등 발현 검정)까지 이어갈 수 있도록 설계했다. 또한, CRAN에 배포함으로써 의존성 관리와 버전 호환성을 보장한다.
실험 결과는 두 가지 주요 축을 중심으로 제시된다. 첫째, 합성 데이터 시뮬레이션을 통해 알려진 세그먼트 경계와 비교했을 때, 평균 절대 오차(MAE)와 Jaccard 지표에서 기존 HMM 기반 혹은 변동점 탐지 방법보다 우수함을 보였다. 둘째, 실제 인간 유전체 RNA‑Seq 데이터셋에 적용해, 알려진 유전자 주석과 비교했을 때, 코딩 영역과 비코딩 영역을 정확히 구분하는 데 높은 재현율과 정밀도를 달성하였다. 특히, 복잡한 전사체 구조(대안 스플라이싱, 중첩 유전자)를 포함한 지역에서도 알고리즘이 안정적으로 작동함을 확인했다.
한계점으로는 φ를 고정값으로 추정하는 과정이 전역적인 과분산을 가정한다는 점이다. 실제 데이터에서는 구간별 분산 차이가 존재할 수 있어, 향후 구간별 φ 추정 혹은 베이지안 계층 모델을 도입하면 더욱 정교한 분할이 가능할 것으로 기대된다. 또한, 현재는 단일 샘플에 대한 분할에 초점을 맞추었지만, 다중 샘플(조건별 비교) 분석을 위한 확장도 필요하다.
결론적으로, 이 논문은 고성능 PDP 알고리즘을 음이항 모델에 성공적으로 적용함으로써, 대규모 시퀀싱 데이터의 정확하고 빠른 구간 탐지를 가능하게 했다. 제공된 R 패키지는 사용 편의성과 재현성을 동시에 만족시키며, 차세대 유전체 분석 파이프라인에 바로 통합될 수 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기