다중 데이터 유형 통합 분석을 위한 JIVE 방법
초록
JIVE(Joint and Individual Variation Explained)는 여러 고차원 데이터 유형을 동시에 분석해 공통(공동) 변동과 각 데이터 유형별 고유 변동을 분리하는 저차원 분해 기법이다. 저순위 근사와 잔차 노이즈를 포함한 3가지 구성요소로 데이터를 표현하며, 기존 PCA, CCA, PLS에 비해 공동·개별 구조를 명확히 구분한다. TCGA의 Glioblastoma 데이터에 적용해 유전자와 miRNA 간 연관성을 밝혀내고, 종양 아형 구분에 기여한다.
상세 분석
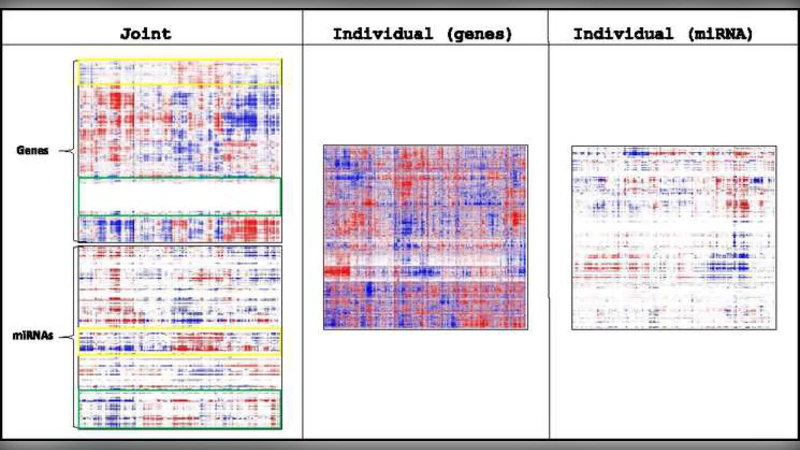
JIVE는 k개의 데이터 행렬 X₁,…,X_k(각 행렬은 동일한 n개의 샘플을 공유) 를 각각 스케일링·중심화한 뒤, 전체 행렬 X를 저차원 공동 구조 J와 개별 구조 A_i 로 분해한다. 수학적으로는 X_i = J_i + A_i + ε_i 로 표현되며, 여기서 J_i는 공동 구조 J의 부분행렬, A_i는 i번째 데이터 유형 전용 저랭크 행렬, ε_i는 독립 잡음이다. 중요한 제약은 J와 A_i 의 행 공간이 서로 직교한다는 점으로, 이는 공동 변동과 개별 변동이 중복되지 않도록 보장한다. 모델 차원(r, r_i)은 퍼뮤테이션 기반 순위 선택 절차를 통해 결정되며, 선택된 차원에 따라 최소제곱 잔차 ‖R‖² 를 최소화하는 교번 알고리즘이 적용된다. 알고리즘은 (1) 현재 J를 고정하고 각 A_i 를 SVD 기반으로 업데이트, (2) 업데이트된 A_i 를 고정하고 J 를 전체 행렬에서 r개의 주성분으로 재추정하는 과정을 반복한다. 이 과정은 잔차 제곱합이 단조 감소함을 보장하므로 수렴성이 확보된다.
JIVE의 핵심 장점은 (i) 데이터 유형 간 스케일 차이를 정규화한 뒤 동일한 가중치로 분석함으로써 “큰 데이터가 우세”하는 현상을 방지하고, (ii) 두 개 이상의 블록을 동시에 다룰 수 있어 다중오믹스 통합에 적합하며, (iii) 공동 구조와 개별 구조를 시각화(히트맵)함으로써 생물학적 해석을 직관적으로 지원한다는 점이다. 논문에서는 GBM(글리오블라스토마) 샘플 234개에 대해 유전자 발현(23,293 변수)과 miRNA 발현(534 변수)을 적용하였다. 퍼뮤테이션 테스트(α=0.01, 1000번) 결과 공동 구조 차원 r=5, 유전자 개별 차원 r₁=33, miRNA 개별 차원 r₂=13이 선택되었다. 공동 구조는 miRNA 변동의 23%, 유전자 변동의 14%를 설명했으며, 유전자 데이터의 58%는 개별 구조로 설명되어 miRNA와는 독립적인 변동임을 확인했다. 히트맵을 통해 특정 유전자·miRNA 군집이 공동 패턴을 공유하고, 다른 군집은 각각의 데이터 유형에 특이적인 패턴을 보였다. 이러한 결과는 miRNA가 유전자 발현을 조절하는 주요 메커니즘 중 하나이지만, 유전자 발현에 영향을 미치는 요인은 다중이며, JIVE가 이를 정량적으로 구분해 줌을 보여준다.
전반적으로 JIVE는 고차원 다중 블록 데이터를 통합 분석할 때, 공동 변동과 개별 변동을 명확히 분리하고, 차원 축소와 시각화를 동시에 제공함으로써 데이터 과학자와 생물학자 모두에게 유용한 도구가 된다.
댓글 및 학술 토론
Loading comments...
의견 남기기