메모리 절감형 희소 k‑mer 그래프 기반 차세대 유전체 어셈블러
초록
**
SparseAssembler2는 기존 de Bruijn 그래프가 요구하는 모든 겹치는 k‑mer을 저장하지 않고, 일정 간격(g)으로 선택된 희소 k‑mer만 보관한다. 선택된 k‑mer 사이에 읽기로부터 추정된 링크를 연결해 그래프를 구성하고, Dijkstra‑유사 너비우선 탐색으로 오류와 다형성을 정제한다. 메모리 사용량은 기존 어셈블러 대비 약 90 % 절감하면서도 정확도와 N50을 유지한다.
**
상세 분석
**
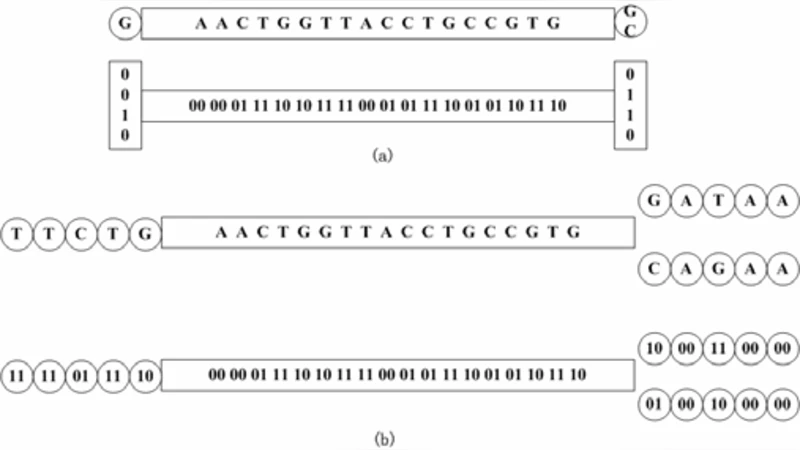
SparseAssembler2는 de Bruijn 그래프의 “모든 k‑mer 저장 → 인접 k‑mer 연결”이라는 기본 구조를 근본적으로 재구성한다. 핵심 아이디어는 전체 k‑mer 집합을 그대로 보관하는 대신, 사용자가 사전에 정의한 간격 g(예: 25)를 기준으로 읽기 스트림을 스캔하면서 g 개의 k‑mer마다 하나를 선택해 노드로 만든다. 이렇게 선택된 노드들은 실제 유전체 상에서 거의 g 베이스씩 떨어져 있기 때문에, 그래프의 정점 수가 원래의 1/g 배 수준으로 감소한다.
노드 선택은 두 단계로 진행된다. 첫 번째 라운드에서는 각 읽기를 순차적으로 탐색하면서 이미 선택된 노드가 나타나면 그 위치로 이동하고, 그렇지 않으면 현재 위치의 k‑mer를 새로운 노드로 등록한다. 두 번째 라운드에서는 선택된 노드들 사이에 읽기로부터 추정된 링크를 구축한다. 이때 각 링크는 앞뒤 k‑mer 사이에 존재하는 g‑1 개의 중간 염기 서열을 압축해 저장한다. 결과적으로 각 노드당 필요로 하는 메모리는 (24 bits + 2 × g bits) 정도이며, 전체 메모리 요구량은
(S \approx \frac{N}{g} \times (24 + 2g) + \text{포인터 오버헤드}) 로 표현된다. 여기서 N은 전체 k‑mer 다양성이다.
오류 정제는 두 단계로 수행된다. 첫 번째는 커버리지가 낮은 노드를 필터링해 스푸리어스 브랜치를 제거하고, 두 번째는 Dijkstra‑유사 너비우선 탐색을 통해 방문한 노드가 재방문될 경우 마지막 분기점으로 되돌아가면서 높은 커버리지를 가진 경로를 보존한다. 이 과정에서 팁(tip)과 버블(bubble) 구조가 효과적으로 소거된다.
SparseAssembler2는 전통적인 de Bruijn 기반 어셈블러가 필요로 하는 대규모 해시 테이블이나 비트맵 압축 구조를 사용하지 않는다. 대신 일반적인 해시 테이블에 희소 k‑mer와 그에 대한 링크 정보를 저장함으로써 구현이 단순하면서도 메모리 효율성을 극대화한다. 실험 결과, 과일파리, 벼, E. coli 등 다양한 규모의 유전체에 대해 기존 어셈블러(SOAPdenovo, Velvet, ABySS)와 비교했을 때 메모리 사용량은 10 % 이하로 감소하고, N50 및 최장 컨티그 길이도 동등하거나 우수한 성능을 보였다. 특히 416 Mbp 꿀벌 게놈 실데이터에서는 메모리 3.5 GB로 SOAPdenovo의 30 GB 대비 10배 이상 절감하면서 N50와 총 조립 길이에서도 소폭 개선을 기록했다.
요약하면, SparseAssembler2는 “희소 k‑mer 선택 + 링크 기반 그래프 탐색”이라는 새로운 모델을 제시함으로써, 메모리 제약이 큰 데스크톱 환경에서도 대형 유전체를 효율적으로 어셈블할 수 있는 실용적인 솔루션을 제공한다.
**
댓글 및 학술 토론
Loading comments...
의견 남기기