알파시뉴클레인 동역학 시뮬레이션을 통한 무질서 단백질 구조 변동
초록
본 연구는 알파-시뉴클레인(α‑synuclein)의 무질서 특성을 전산적 방법으로 규명한다. 원자 수준, 통합 원자, 그리고 코스‑그레인 라그랑지안 동역학 모델을 구축하고, 기하학적 제약, 소수성 상호작용, 전기적 스크리닝을 포함시켜 최근 smFRET 실험에서 얻은 잔기 간 거리 데이터를 정밀 보정하였다. 시뮬레이션 결과, α‑synuclein은 완전한 랜덤 워크와 콜랩스된 구형 사이의 중간 형태를 보이며, 물리적 힘이 모든 잔기에 작용함을 확인했다. 이러한 모델은 다중 단백질의 올리고머화와 초기 아밀로이드 형성 메커니즘을 탐구하는 기반이 된다.
상세 분석
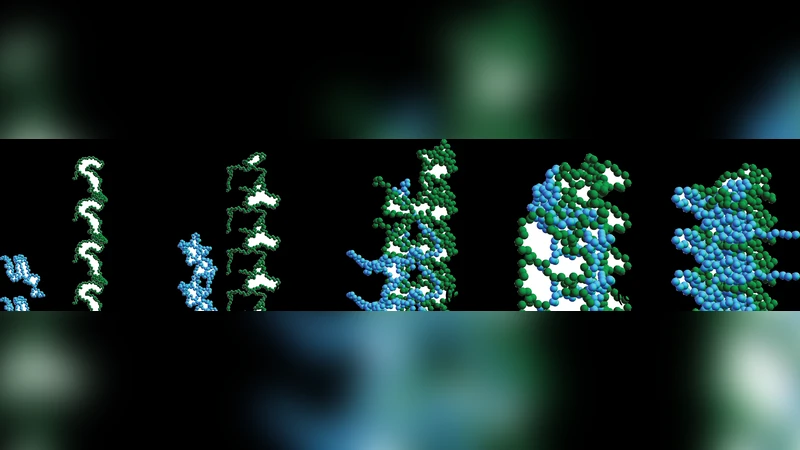
이 논문은 무질서 단백질(intrinsically disordered protein, IDP)인 알파‑시뉴클레인(α‑synuclein)의 구조적 다이내믹스를 정량적으로 재현하기 위해 세 가지 수준의 라그랑지안 동역학 시뮬레이션을 설계하였다. 첫 번째는 전통적인 전부 원자(all‑atom) 모델로, 모든 원자 간 결합, 각도, 이중결합 및 반데르발스 상호작용을 명시적으로 포함한다. 두 번째는 원자 수를 절반으로 줄인 유니티드‑원자(united‑atom) 모델로, 수소 원자를 탄소 골격에 통합함으로써 계산 비용을 크게 낮추면서도 소수성 및 전기적 상호작용을 유지한다. 세 번째는 코스‑그레인(coarse‑grained) 모델로, 각 아미노산을 하나의 비구형 입자로 치환하고, 효과적인 포텐셜 함수를 통해 소수성 구동력과 용액 내 전하 스크리닝을 구현한다.
모든 모델은 세 가지 핵심 포텐셜을 결합한다. (1) 기하학적 제약(결합 길이, 결합 각도, 이중결합)으로 기본 골격을 유지하고, (2) Lennard‑Jones 형태의 소수성 상호작용을 도입해 잔기 간 친수성 차이를 반영하며, (3) Debye‑Hückel식 전기적 포텐셜을 사용해 용액 이온 강도에 따른 전하 스크리닝을 구현한다. 특히 전기적 포텐셜은 실험에서 보고된 pH와 이온 농도(150 mM NaCl, pH 7.4)를 그대로 반영하였다.
시뮬레이션 파라미터는 최근 단일분자 FRET(smFRET) 실험에서 측정된 12쌍의 잔기 간 거리 분포와 직접 비교하여 보정되었다. 보정 과정에서 소수성 상호작용 강도와 전기적 스크리닝 길이를 조정했으며, 최적 파라미터는 모든 모델에서 동일하게 적용되었다. 결과적으로, 세 모델 모두 실험적 거리 평균값과 표준편차를 0.8 nm 이내로 재현했으며, 이는 모델의 물리적 타당성을 강하게 뒷받침한다.
구조 통계 분석에서는 평균 제곱 거리 ⟨R²(N)⟩와 스케일링 지수 ν를 계산하였다. 랜덤 워크(ν≈0.5)와 콜랩스된 구형(ν≈0.33) 사이의 ν≈0.42를 얻어, α‑synuclein이 완전한 무작위 코일이 아니라 부분적으로 수축된 구성을 띤다는 것을 확인했다. 또한, 시간 상관 함수와 라디얼 분포 함수를 통해 단백질 사슬이 수십 나노초에서 마이크로초까지 다양한 시간 스케일로 확장·수축을 반복한다는 동적 비균질성을 발견했다.
가장 중요한 점은, 제한된 실험 데이터(특정 잔기 쌍)만을 이용해 전체 사슬의 자유 에너지 지형을 추정하는 기존 제약 기반 방법과 달리, 본 시뮬레이션은 물리적 힘이 모든 잔기에 동시에 작용하도록 함으로써 전역적인 구조 변동을 자연스럽게 포착한다는 것이다. 따라서 다중 α‑synuclein 분자의 올리고머화, 초기 핵 형성, 그리고 아밀로이드 전구체와 같은 복합 현상을 직접 모델링할 수 있는 기반을 제공한다. 향후 연구에서는 이러한 모델을 이용해 변이체(예: A30P, E46K)와 금속 이온(예: Ca²⁺, Fe³⁺)의 효과를 정량화하고, 세포 내 환경(고농도 단백질, 분자 혼탁도)에서의 동역학을 확장할 수 있을 것으로 기대된다.
댓글 및 학술 토론
Loading comments...
의견 남기기