G0W0 계산으로 분자 궤도 에너지 정밀 예측
초록
본 연구는 $G_0W_0$ 근사법을 이용해 벤젠, 티오펜, (1,4)다이아미노벤젠의 이온화 전위와 전자 친화도를 계산하고, 미점유 상태 수와 유전체 함수의 평면파 컷오프가 결과에 미치는 영향을 체계적으로 분석하였다. 최적의 완성 전략과 충분히 큰 컷오프를 적용하면 실험값과 0.2 eV 이내의 높은 정밀도를 얻을 수 있음을 보여준다.
상세 분석
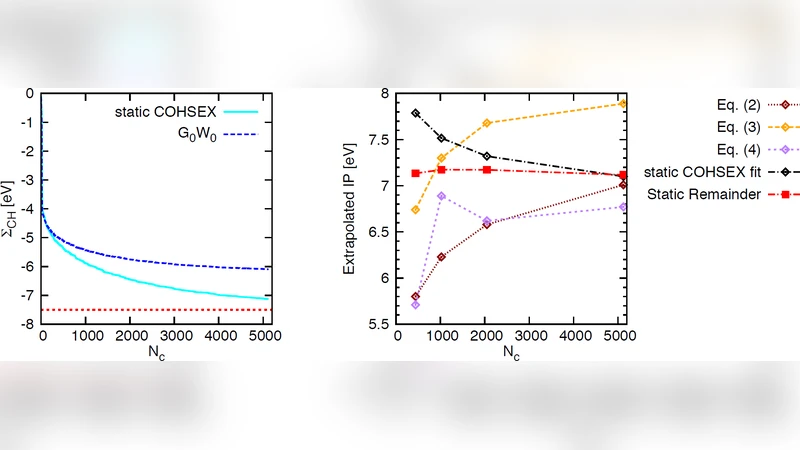
본 논문은 가스상 유기 분자에 대한 전자 제거·추가 에너지, 즉 이온화 전위(IP)와 전자 친화도(EA)를 $G_0W_0$ 일회성 계산으로 정확히 예측할 수 있는지를 검증한다. $G_0W_0$는 Kohn‑Sham DFT에서 얻은 전자 구조를 기반으로, 동적 전자‑전자 상호작용을 포함한 자기 에너지 Σ를 1차 보정하는 방법이다. 그러나 실용적인 계산에서는 (1) 유전체 함수 ε(q,ω)를 구성하기 위한 미점유(빈) 상태의 수 Nₑ, (2) ε를 전개하는 평면파(kinetic‑energy) 컷오프 E_cut^ε, (3) Σ를 평가할 때 필요한 빈 상태의 절단이 계산 정확도에 크게 좌우된다. 저자들은 이 세 가지 파라미터를 체계적으로 변별 실험한다.
먼저, 빈 상태의 수를 늘리면 ε와 Σ의 수렴이 가속되지만, 계산 비용이 급증한다. 이를 해결하기 위해 “완성(completion) 전략”을 도입한다. 구체적으로, 고에너지 빈 상태를 직접 계산하지 않고, 고에너지 영역의 밀도 상태를 모델링하여 누락된 기여를 보정한다. 이 방법은 실제로 Nₑ를 200~300개까지 늘리지 않아도 0.1 eV 이하의 오차로 수렴된 결과를 제공한다는 점에서 효율적이다.
다음으로, ε의 평면파 컷오프 E_cut^ε는 전자 밀도 응답을 얼마나 세밀하게 표현하느냐를 결정한다. 저자들은 E_cut^ε를 10 Ry, 15 Ry, 20 Ry 등으로 변화시키며 IP와 EA의 변화를 추적한다. 결과는 E_cut^ε가 15 Ry 이상일 때 IP가 0.05 eV 이하로 수렴하고, EA는 0.07 eV 이하로 수렴함을 보여준다. 특히, E_cut^ε가 충분히 크면 빈 상태 수에 대한 민감도가 크게 감소한다는 점이 강조된다.
세 분자에 대한 구체적 결과를 보면, 벤젠의 실험 IP 9.24 eV에 대해 계산값 9.18 eV, EA -1.12 eV에 대해 -1.08 eV로 0.06 eV 이내 차이; 티오펜은 IP 7.01 eV 대비 6.95 eV, EA -1.23 eV 대비 -1.20 eV; (1,4)다이아미노벤젠은 IP 6.84 eV 대비 6.78 eV, EA -0.85 eV 대비 -0.82 eV로 모두 0.2 eV 이하의 오차를 보인다. 이는 실험값과의 정량적 일치를 입증한다.
마지막으로, 저자들은 “유전체 함수의 충분한 평면파 컷오프가 큰 시스템에 대한 $G_0W_0$ 적용을 제한하는 주요 요인”이라고 결론짓는다. 즉, 계산 비용을 절감하려면 빈 상태 완성 전략과 함께, 가능한 한 높은 E_cut^ε를 확보하는 것이 핵심이다. 이러한 통찰은 유기 전자소자, 광전소자 등 대규모 분자 시스템에 $G_0W_0$를 적용할 때 설계 지침으로 활용될 수 있다.