DNA 결합 단백질 예측을 위한 효율적인 머신러닝 접근
초록
본 논문은 전체 전하, 패치 크기, 아미노산 조성 등 3가지 물리·화학적 특성을 이용해 DNA 결합 단백질을 구분하는 모델을 구축한다. 지원 벡터 머신(SVM)과 계단식 상관 신경망(CCNN)을 각각 최적화하고, 잭나이프 교차검증으로 성능을 비교하였다. SVM(ANOVA 커널)은 정확도 86.7%, 민감도 91.1%, 특이도 85.3%를 기록했으며, CCNN은 정확도 75.4%, 민감도 82.6%, 특이도 72.3%에 머물렀다.
상세 분석
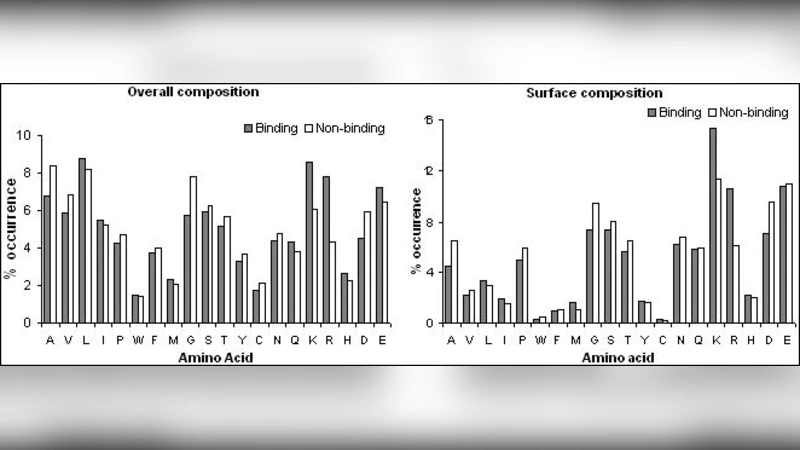
이 연구는 DNA‑binding protein(DNA‑BP) 예측이라는 생물정보학 문제에 머신러닝을 적용한 전형적인 사례로, 데이터셋 구성, 피처 설계, 모델 선택, 평가 방법론이 체계적으로 제시된다. 먼저 121개의 DNA‑BP와 238개의 비결합 단백질을 수집했으며, 이는 기존 연구에서 사용된 데이터와 비교했을 때 비교적 균형 잡힌 샘플링을 보여준다. 피처는 전체 전하(overall charge), 패치 크기(patch size), 아미노산 조성(amino‑acid composition)이라는 세 가지 물리·화학적 속성을 기반으로 하며, 각각이 단백질‑DNA 상호작용에 미치는 영향을 기존 문헌에서 뒷받침한다. 전하와 패치 크기는 전자적 친화도와 결합 부위의 공간적 특성을 반영하고, 아미노산 조성은 서열 수준에서의 친화성을 포착한다.
모델링 단계에서 저자는 두 가지 학습 알고리즘을 선택했다. 첫 번째는 커널 기반 분류기인 SVM이며, 특히 ANOVA 커널을 사용해 비선형 관계를 효과적으로 모델링한다. ANOVA 커널은 다차원 입력 공간을 다항식 형태로 변환하면서 차원 저주 현상을 완화시켜, 제한된 데이터에서도 과적합을 방지한다. 하이퍼파라미터(C, γ 등)는 그리드 탐색과 교차검증을 통해 최적화되었으며, 최종 모델은 잭나이프(leave‑one‑out) 방식으로 평가되어 일반화 성능을 신뢰성 있게 추정한다.
두 번째는 계단식 상관 신경망(CCNN)이다. CCNN은 기존 신경망에 비해 구조가 동적으로 성장하면서 새로운 은닉 유닛을 추가해 학습 데이터를 점진적으로 설명한다. 이는 작은 데이터셋에서도 복잡한 비선형 패턴을 포착할 수 있다는 장점이 있다. 그러나 본 연구에서는 전체 데이터를 사용해 최적화했음에도 불구하고, SVM에 비해 정확도와 특이도가 현저히 낮았다. 이는 CCNN이 파라미터 수가 많아 과적합 위험이 크고, 피처 차원이 제한적일 때 충분한 표현력을 발휘하지 못했을 가능성을 시사한다.
성능 평가는 정확도, 민감도(Recall), 특이도(Specificity) 세 가지 지표로 이루어졌으며, 특히 민감도가 91.1%에 달한 SVM은 DNA‑BP를 놓치지 않는 능력이 뛰어나다는 점을 강조한다. 반면 특이도가 85.3%로 완벽하지 않아, 비결합 단백질을 오분류하는 경우가 존재한다. 이는 실제 생물학적 응용에서 false positive를 최소화하기 위한 추가적인 후처리나 다중 모델 앙상블이 필요함을 암시한다.
전체적으로 이 논문은 제한된 피처와 데이터에도 불구하고 SVM이 강력한 분류 성능을 보임을 입증한다. 또한, 커널 선택과 교차검증 전략이 결과에 미치는 영향을 명확히 제시함으로써 향후 DNA‑BP 예측 연구에 대한 벤치마크로 활용될 수 있다. 향후 연구에서는 구조 기반 피처(예: 3D 접촉 지도)와 딥러닝 기반 자동 피처 추출을 결합해 성능을 더욱 향상시킬 여지가 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기