DNA 혼합 분석을 위한 MAIES 전문가 시스템
초록
MAIES는 정량적 피크 면적 정보를 조건부 가우시안 분포로 모델링하고, 정밀한 정점 트리 전파를 통해 DNA 혼합물에 포함된 기여자를 식별하거나 미지 기여자의 프로파일을 예측하는 전문가 시스템이다. 실제 사례를 통해 MAP 탐색 알고리즘의 효율성을 입증하였다.
상세 분석
MAIES는 법의학 DNA 혼합 분석이라는 복합 문제를 해결하기 위해 베이지안 네트워크와 정확한 확률 추론 기법을 결합한 독창적인 프레임워크를 제시한다. 핵심 아이디어는 각 피크의 면적을 연속형 변수로 취급하고, 이를 조건부 가우시안 분포로 표현함으로써 정량적 정보를 손실 없이 모델링한다는 점이다. 이러한 연속형 변수는 전통적인 이산형 마커 기반 시스템에서 다루기 어려운 불확실성을 자연스럽게 포착한다.
네트워크 구조는 기여자별 유전형 변수와 관측된 피크 면적 변수 사이의 의존성을 명시적으로 정의한다. 각 기여자는 두 개의 대립 유전자를 갖는 다중 대립형질을 가지고 있으며, 이들 유전형은 Hardy‑Weinberg 평형과 독립적인 유전자 흐름을 가정한다. 관측 변수는 해당 유전형이 생성할 수 있는 피크 면적의 평균값과 분산을 기반으로 조건부 가우시안으로 연결된다.
추론 단계에서는 정확한 정점 트리(junction tree) 전파 알고리즘을 적용한다. 먼저 베이지안 네트워크를 삼각화하고 클리크 트리를 구성한 뒤, 클리크 간에 메시지를 교환하여 마진 분포와 사후 확률을 계산한다. 이 과정은 연속형 변수와 이산형 변수가 혼합된 하이브리드 모델에서도 수치적 적분 없이 정확한 결과를 제공한다는 장점이 있다.
MAIES의 핵심 기능 중 하나는 MAP(최대 사후 확률) 탐색 알고리즘이다. 논문 부록에 제시된 알고리즘은 탐색 공간을 효율적으로 축소하기 위해 기여자 수와 가능한 유전형 조합을 단계별로 제한한다. 구체적으로, 초기 단계에서 관측된 피크 면적과 일치하는 유전형 후보군을 필터링하고, 이후 클리크 트리에서 얻은 사후 확률을 이용해 후보군의 순위를 매긴다. 이렇게 하면 전체 조합 수가 기하급수적으로 증가하는 문제를 회피하면서도 최적 해에 근접한 결과를 얻을 수 있다.
시스템 구현 측면에서 MAIES는 C++ 기반의 추론 엔진과 사용자 친화적인 GUI를 결합하였다. 입력 데이터는 표준 STR(Short Tandem Repeat) 프로파일과 피크 면적 값으로 구성되며, 시스템은 자동으로 데이터 정규화와 전처리를 수행한다. 결과 출력은 기여자별 사후 확률, 가능한 프로파일 리스트, 그리고 혼합물 분리 결과를 시각적으로 제공한다.
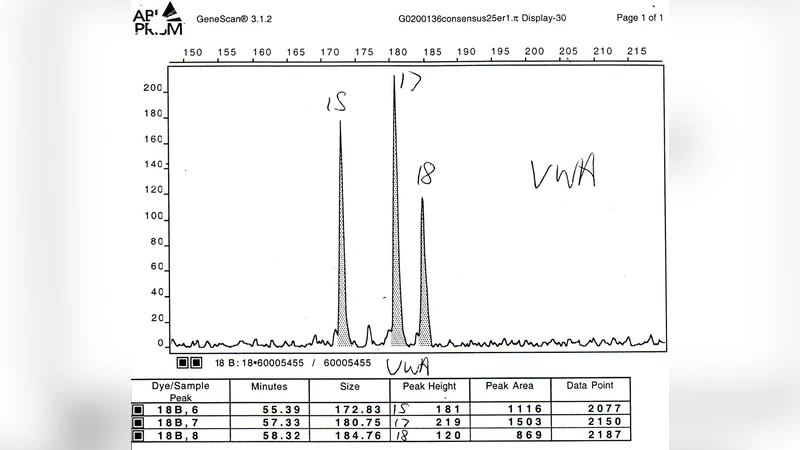
실제 사례 적용에서는 두 명의 알려진 기여자와 하나의 미지 기여자가 포함된 혼합물을 분석하였다. MAIES는 알려진 기여자를 정확히 식별하고, 미지 기여자의 프로파일을 높은 확률로 예측하였다. 특히, 피크 면적이 겹치는 복잡한 구간에서도 조건부 가우시안 모델이 효과적으로 불확실성을 반영하여 정확도를 유지한 점이 주목할 만하다.
한계점으로는 가우시안 가정이 실제 피크 면적 분포와 완전히 일치하지 않을 수 있다는 점과, 매우 많은 기여자가 존재하는 경우 클리크 트리의 크기가 급증해 계산 비용이 증가한다는 점을 들 수 있다. 향후 연구에서는 비가우시안 혼합 모델이나 근사 추론 기법을 도입해 확장성을 높이는 방안을 제시한다.
전반적으로 MAIES는 정량적 DNA 혼합 분석에 베이지안 추론을 적용한 최초의 시스템 중 하나로, 정확한 확률적 결론을 제공함으로써 법의학 현장에서 증거 해석의 신뢰성을 크게 향상시킬 수 있다.