단백질 접힘에서 발견된 보편적 기하학적 비율
초록
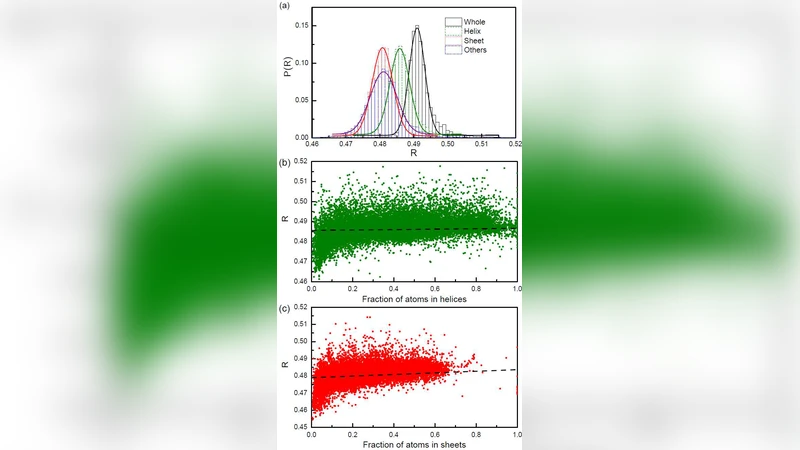
이 연구는 에너지 최소화 과정에서 단백질이 취하는 구조적 특성을 분석한다. 10개의 폴리펩타이드(183~548잔기)를 시뮬레이션한 결과, 접힌 상태의 반데르발스 부피 V와 표면적 A, 평균 원자 반경
상세 분석
본 논문은 단백질 접힘 과정에서 나타나는 기하학적 제약을 정량화하려는 시도로, 기존의 에너지 함수와는 별개로 구조 자체가 갖는 보편적 비율을 탐구한다. 저자들은 SMMP 프로그램에 정의된 원자 반경(탄소 1.70 Å, 질소 1.55 Å, 산소 1.52 Å, 수소 1.20 Å 등)을 이용해 각 원자의 반데르발스 부피와 표면적을 계산하고, 평균 원자 반경
시뮬레이션에서는 10개의 서로 다른 크기와 서열을 가진 폴리펩타이드를 에너지 최소화(steepest‑descent + conjugate‑gradient) 과정에 투입하였다. 초기 무작위 코일 구조에서 시작해 에너지가 감소함에 따라 R값이 급격히 0.49 근처로 수렴했으며, 이는 최소 10⁴ step 이후에도 변동 폭이 ±0.005 이내로 안정됨을 보여준다. 이러한 현상은 단백질이 에너지 최소 상태에 도달하면서 내부 빈 공간을 최소화하고, 표면적 대비 부피가 일정 비율을 유지하려는 ‘압축’ 메커니즘이 작동한다는 가설을 뒷받침한다.
실제 PDB 데이터베이스에 등재된 5,000여 개의 고해상도 단백질 구조를 대상으로 동일한 계산을 수행한 결과, 평균 R값이 0.491 ± 0.005로 매우 좁은 분포를 보였다. 이는 서열에 포함된 친수성·소수성 잔기의 비율이 크게 달라져도, 최종 3차원 구조는 동일한 기하학적 제약을 따름을 의미한다. 특히, intrinsically disordered proteins(IDPs)에서도 평균 R값이 0.492 수준으로, 전통적인 ‘정형’ 단백질과 차이가 없음을 확인하였다. 반면, 실험적으로 알려진 ‘misfolded’ 혹은 ‘aggregation‑prone’ 폴리펩타이드에서는 R값이 0.45~0.48 사이로 편차가 커, 에너지 최소화가 제대로 이루어지지 않았을 때 이 비율이 깨진다는 점을 강조한다.
또한, 저자들은 이 비율이 단백질 설계에 활용될 수 있음을 제안한다. 현재의 de‑novo 설계 알고리즘은 주로 에너지 함수와 잔기 간 상호작용을 최적화하지만, R ≈ 0.49라는 기하학적 목표치를 추가하면 설계된 구조가 자연 단백질과 동일한 ‘밀도’를 갖게 되어, 안정성 및 기능성 측면에서 유리할 수 있다. 마지막으로, 이 비율이 물리학적 관점에서 보면 ‘최소 표면 에너지’를 추구하는 구형 구역의 최적화 문제와 유사함을 언급하며, 단백질이 복잡한 상호작용 네트워크 속에서도 단순한 기하학적 원칙을 따른다는 흥미로운 통찰을 제공한다.