질병 경로 프로파일링 변동성 분석 플랫폼
초록
본 논문은 질병 특이적 경로 모델을 도출하고, 전역 구조 변화와 개별 링크 파괴를 정량화할 수 있는 네트워크 지표들을 제시한다. 네트워크 비교와 머신러닝 기반 분자 프로파일링을 결합한 오픈소스 파이프라인을 설계하고, 분류기, 특징 선택·정렬, 풍부도 분석, 네트워크 추론 및 비교 함수 등 다섯 가지 주요 변동 요인을 체계적으로 평가한다. 파킨슨병 후기 단계 마이크로어레이 데이터를 대상으로 다양한 머신러닝 방법을 적용한 결과, 서로 다른 경로 프로파일링 결과가 낮은 겹침을 보였지만 각각 의미 있는 생물학적 인사이트를 제공함을 확인하였다.
상세 분석
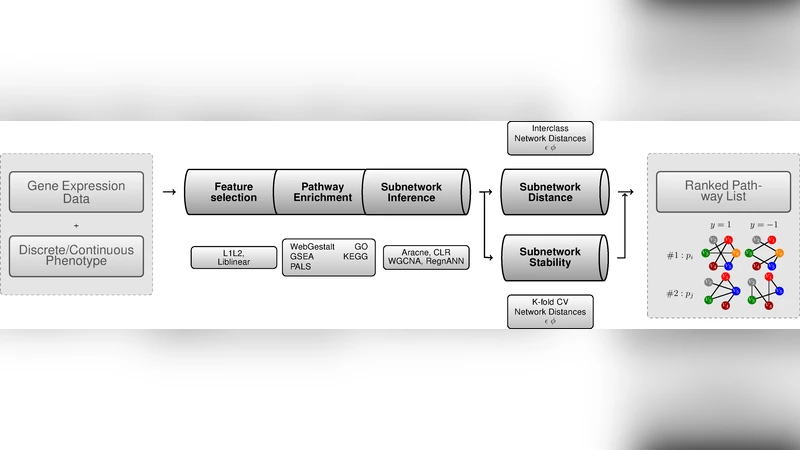
이 연구는 ‘경로 프로파일링’이라는 개념을 네트워크 과학과 머신러닝을 융합함으로써 새로운 차원으로 확장한다. 먼저, 원시 유전자 발현 데이터를 기반으로 여러 가지 특징 선택·정렬 알고리즘(예: t‑test, LIMMA, Random Forest 중요도 등)을 적용하고, 선택된 유전자 집합을 KEGG·Reactome 등 기존 데이터베이스와 매핑해 경로 풍부도 분석을 수행한다. 여기서 핵심은 동일한 데이터에 대해 서로 다른 선택·정렬 전략이 전혀 다른 경로 집합을 도출한다는 점이다. 이는 ‘분석 파이프라인의 변동성’이 결과 해석에 큰 영향을 미칠 수 있음을 시사한다.
다음 단계에서는 선택된 경로를 기반으로 네트워크를 재구성한다. 논문은 두 가지 주요 네트워크 추론 방식을 제시한다. 첫째는 상관 기반 가중 네트워크(Weighted Gene Co‑expression Network Analysis, WGCNA)이며, 둘째는 베이지안 네트워크와 같은 확률적 그래프 모델이다. 각 방법은 노드(유전자)와 엣지(상호작용)의 정의가 다르기 때문에, 동일한 경로라도 네트워크 토폴로지가 크게 달라진다.
네트워크 비교 단계에서는 전역 구조 차이를 측정하기 위해 그래프 직경, 클러스터링 계수, 모듈러티 등 전통적인 지표와, 특정 엣지의 존재·부재를 평가하는 ‘링크 교체 점수(link replacement score)’를 도입한다. 이러한 다중 지표는 한 방법이 놓친 미세한 변화를 다른 방법이 포착하도록 설계되었다.
마지막으로, 머신러닝 분류기(예: SVM, Random Forest, Logistic Regression)를 이용해 환자군과 대조군을 구분한다. 여기서 중요한 점은 동일한 특징 집합이라도 분류 알고리즘에 따라 성능이 크게 변한다는 것이다. 논문은 교차 검증과 부트스트랩을 통해 각 단계별 변동성을 정량화하고, 최종 결과가 ‘다양한 방법을 통합했을 때 가장 풍부하고 신뢰성 있는 생물학적 해석을 제공한다’는 결론에 도달한다.
전체 파이프라인은 Docker와 Snakemake를 활용해 완전한 재현성을 보장하도록 구현되었으며, 모든 스크립트와 의존성은 GitHub에 오픈소스로 공개된다. 이러한 투명한 구현은 향후 다른 질병 데이터셋에 대한 확장과 비교 연구를 용이하게 만든다.
댓글 및 학술 토론
Loading comments...
의견 남기기