자동 RNA 구조 예측으로 밝혀진 이중 글리신 리보스위치의 숨겨진 연결 고리
초록
이 연구는 최신 자동 RNA 3차원 구조 예측 도구 네 가지를 이용해 이중 글리신 리보스위치의 5′·3′ 플랭킹 영역에 존재하던 미지의 P0 줄기와 켁‑턴(kink‑turn) 모티프를 발견하고, 화학적 매핑·돌연변이·구조 모델링을 통해 이 구조가 리보스위치의 두 단계 글리신 결합에 필수적인 에너지 기여(≈4.3 kcal/mol)를 한다는 것을 실험적으로 입증하였다.
상세 분석
본 논문은 기능성 RNA의 3차원 구조를 자동화된 알고리즘으로 예측하고, 이를 실험적으로 검증하는 최초의 사례 중 하나이다. 저자들은 360개의 이중 글리신 리보스위치 서열을 5′·3′ 플랭킹 영역까지 100 nt씩 확장한 다중 정렬을 구축하고, 네 가지 독립적인 도구—신규 개발된 BPPalign, RMdetect, JAR3D, Rosetta/FARFAR—를 적용하였다. BPPalign은 동종 서열군에서 염기쌍 확률을 평균화해 새로운 줄기(P0)를 제시했고, RMdetect와 JAR3D는 이 영역에 전형적인 켁‑턴 서열·구조 특징을 강하게 탐지하였다. Rosetta 기반 3D 모델링은 P0와 기존 P1 사이에 3‑nt bulge가 존재함을 전제로, 켁‑턴이 두 aptamer를 매끄럽게 연결하면서 전반적인 헬리컬 인터페이스를 형성하도록 재구성하였다. 특히, 켁‑턴의 골격이 기존 결정구조(4.4 Å C4′ RMSD)와 거의 일치한다는 점은 모델링 정확성을 뒷받침한다.
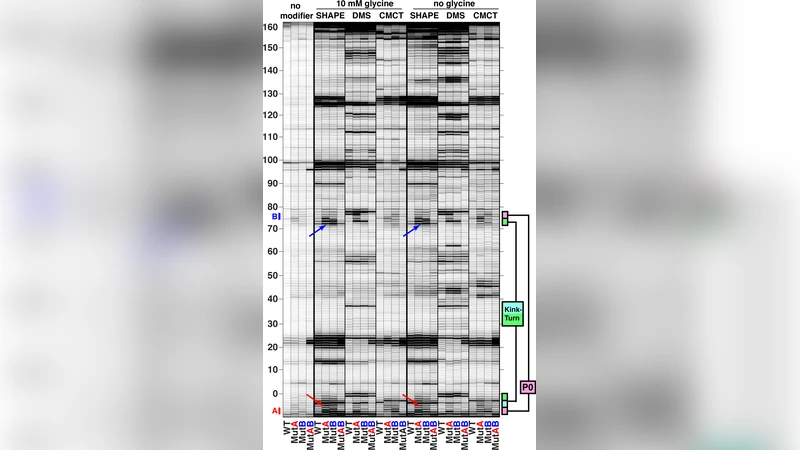
실험적 검증은 화학적 매핑(SHAPE, DMS, CMCT)과 돌연변이‑매핑(mutate‑and‑map), 돌연변이‑구조 회복(mutant rescue)으로 수행되었다. 5′ 플랭킹을 포함한 FN‑KTtest RNA를 설계해, P0 줄기의 Watson‑Crick 쌍을 파괴한 MutA·MutB 변이에서 해당 영역과 인접 P1 퓨린‑퓨린 쌍이 크게 노출되어 SHAPE 신호가 증가하였다. 두 변이를 동시에 복원한 MutAB는 원래와 동일한 보호 패턴을 회복함으로써 구조적 연관성을 명확히 했다.
에너지 측면에서는 글리신 결합 전이를 두 단계(K1, K2)로 구분해 DMS 반응성을 정량화하였다. Wild‑type은 K1≈9 µM, K2≈1.8 mM였으며, MutA·MutB는 각각 K1이 120‑배, K2가 6‑30배 감소하였다. 이는 P0·케크‑턴이 첫 번째와 두 번째 글리신 결합에 각각 2.8 kcal/mol, 1.5 kcal/mol의 자유에너지 기여를 함을 의미한다. MutAB 복원 시 원래 결합 상수로 회복돼 구조‑기능 연관성이 확증되었다.
이러한 결과는 기존 7년간의 연구에서 과도하게 절단된 RNA 서열이 실제 기능에 필수적인 구조 요소를 숨기고 있었음을 보여준다. 또한, 플랭킹 영역에 추가적인 미지의 모티프가 존재할 가능성을 제시하며, 자동화된 구조 예측이 아직 탐색되지 않은 RNA 기능 영역을 발굴하는 강력한 도구가 될 수 있음을 입증한다.
댓글 및 학술 토론
Loading comments...
의견 남기기