비모수 베이지안 희소 요인 모델을 이용한 유전자 발현 분석
초록
이 논문은 인디언 뷔페 프로세스(IBP)를 희소성 priors 로 활용해 잠재 요인의 수를 자동 추정하는 비모수 베이지안 팩터 분석 모델을 제안한다. 관측된 유전자 발현 행렬 Y를 무한히 많은 숨은 요인 X와 가중치 행렬 G의 선형 결합으로 표현하고, G에 IBP를 적용해 대부분의 요소가 0이 되도록 강제한다. 모델은 합성 데이터와 실제 E. coli 및 인간 세포 데이터에 대해 실험적으로 검증되며, 기존 방법 대비 더 정확한 연결 구조 복원과 차원 추정 능력을 보인다.
상세 분석
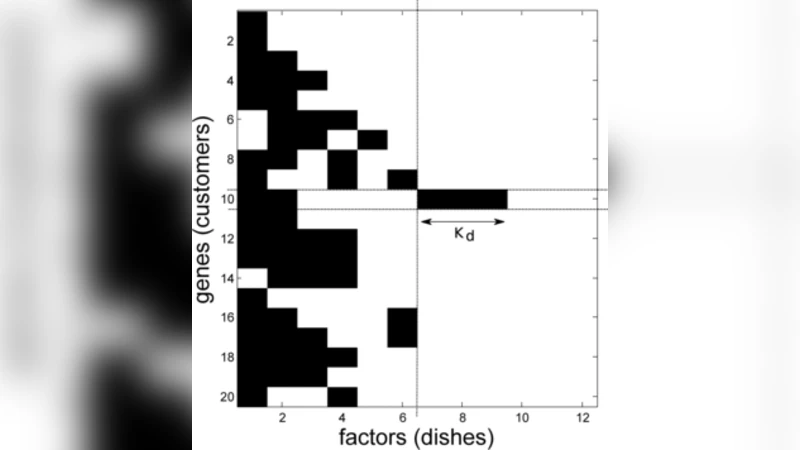
본 연구는 전통적인 팩터 분석(Factor Analysis, FA)이 사전에 지정된 요인 수와 밀집된 로딩 행렬을 가정한다는 한계를 극복하고자, 비모수 베이지안 접근법을 도입한다. 핵심 아이디어는 무한히 많은 잠재 요인을 가정하되, 실제 데이터에 기여하는 요인만 선택적으로 활성화하는 것이다. 이를 위해 인디언 뷔페 프로세스(Indian Buffet Process, IBP)를 G, 즉 요인-특징 매트릭스에 대한 사전 분포로 채택한다. IBP는 각 관측 샘플이 무한히 많은 이진 특성을 가질 확률을 정의하며, ‘희소성’이라는 자연스러운 제약을 제공한다. 즉, 대부분의 요인-특징 연결이 0이 되고, 소수의 강한 연결만 남는다.
모델 수식은 Y = G X + E 로 표현되며, 여기서 X는 연속형 잠재 요인 행렬, E는 가우시안 잡음이다. G는 이진 행렬(IBP에 의해 생성)과 실수형 가중치 행렬의 곱으로 구성될 수 있다. 베이지안 추론은 마르코프 체인 몬테 카를로(MCMC) 방법, 특히 Gibbs 샘플링과 메트로폴리스-헤이스팅스 스텝을 결합해 수행된다. 샘플링 과정에서 요인 수는 자동으로 조정되며, 불필요한 요인은 사후 확률이 0에 수렴한다.
실험 설계는 두 단계로 나뉜다. 첫 번째는 E. coli 대사 네트워크에서 알려진 희소 연결 구조를 기반으로 합성 데이터를 생성해 모델이 원래 구조를 복원할 수 있는지를 평가한다. 두 번째는 실제 유전자 발현 데이터셋(소규모, 중규모, 대규모)에서 모델의 차원 추정 정확도와 예측 성능을 기존 PCA, 전통적 FA, 스파스 코딩 기반 방법과 비교한다. 결과는 IBP 기반 모델이 요인 수를 과소·과대 추정하지 않고, 재현된 연결 매트릭스가 실제 생물학적 네트워크와 높은 상관성을 보임을 보여준다. 또한, 잡음이 큰 상황에서도 모델은 안정적인 잠재 구조를 학습한다.
이 논문의 주요 기여는 (1) 비모수 베이지안 프레임워크를 팩터 분석에 적용해 요인 수를 데이터에 의해 자동 결정하도록 한 점, (2) IBP를 통해 자연스러운 희소성을 부여함으로써 유전자 발현과 같은 고차원·저샘플 데이터에 적합한 모델을 제시한 점, (3) 다양한 실험을 통해 모델의 실용성을 입증한 점이다. 특히, 생물학적 해석 가능성을 높이는 ‘희소 연결’ 특성은 네트워크 재구성, 기능 유전자 탐색 등에 직접 활용될 수 있다. 향후 연구에서는 변분 추론을 통한 스케일업, 다른 비정형 데이터 타입에 대한 확장, 그리고 사전 하이퍼파라미터 자동 튜닝 메커니즘을 도입할 여지가 있다.
댓글 및 학술 토론
Loading comments...
의견 남기기